Northwestern Medicine scientists have identified the role of key architectural proteins in the regulating the expression of the gene that is mutated in cystic fibrosis (CF).
The findings, published in the journal Nucleic Acids Research, shed light on the mechanisms controlling the gene, which underlies this life-threatening disease. CF affects 30,000 children and adults in the United States.
The discovery of the role of these architectural proteins could lead to novel drug therapies for some cystic fibrosis patients, says Ann Harris, PhD, professor in Pediatrics-Human Molecular Genetics at Northwestern University Feinberg School of Medicine.
“This finding is significant because for the first time we have a handle on proteins that can increase CF gene expression in many cell types. This provides new opportunities to target gene expression pathways for novel therapeutics,” said Harris, who is also program director of Human Molecular Genetics Program at Stanley Manne Children’s Research Institute.
It is widely known that mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene cause cystic fibrosis. Mutations in the gene result in lower levels of gene expression or the production of a defective protein by the same name. This is associated with thick, sticky mucus that clogs the lungs, leads to dangerous infections, and can also prevent the body from breaking down food and absorbing nutrients.
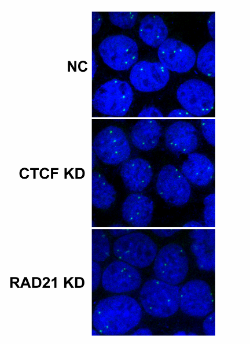
Northwestern scientists, led by the paper’s first author Nehal Gosalia, ’14 PhD, previously observed two architectural proteins – CTCF and the Cohesin Complex – actively binding at sequences near the gene in epithelial cells, which are the site of CF disease.

In normal genes, says Gosalia, these proteins have important roles in the establishment and maintenance of gene expression – the process by which genes manufacture certain proteins. However, using chromosome conformation capture techniques and fluorescence microscopy, Gosalia showed that at the CFTR gene, the two architectural proteins have distinct, but critical roles in regulating the organization of the gene; with one acting as a mediator of interactions between regulatory sequences and the other as a stabilizer of the 3D structure of the active gene. Part of this work was a collaboration with graduate student, Daniel Neems in the lab of Steven Kosak, PhD, assistant professor in Cell and Molecular Biology.
“We showed that these architectural proteins are responsible for fine-tuning the production of the cystic fibrosis gene mRNA through the maintenance and organization of its structure,” Gosalia said. “They are also involved in regulating the genetic landscape and the positioning of the gene in the nucleus.”
The information learned about these proteins at the CFTR gene could be used to create better therapeutics.
“Current drug therapies for cystic fibrosis target the defective protein, but not all patients have enough of this protein in their bodies to correct,” said Gosalia. “We want to better understand the process by which this gene is expressed, so it may lead to better drug therapies.”
The research was funded by the Cystic Fibrosis Foundation, National Institutes of Health grants R01 HL094585, and HD068901.