Modulating the activity of a kinase in motor neurons may help mitigate mitochondrial defects and other symptoms of spinal muscular atrophy, offering a new therapeutic avenue for the devastating disease, according to a Northwestern Medicine study published in the Proceedings of the National Academy of Sciences (PNAS).
Spinal muscular atrophy (SMA), a genetic disease affecting motor neurons, disrupts the nerve cells that control voluntary muscle movement, according to the Muscular Dystrophy Association. Children born with SMA typically have an impaired ability to crawl, walk, sit up and control their head movements.
The disease is the top genetic cause of toddler mortality, as most children born with type 1 SMA will die before their second birthday, according to the National Institutes of Health.

Despite the debilitating effects of the disease, the molecular mechanisms that trigger the development of SMA are not well understood, said senior study author Yongchao C. Ma, PhD, associate professor of Pediatrics, of Neuroscience, and in the Ken and Ruth Davee Department of Neurology.
“SMA is a pediatric motor neuron disease; It’s almost like ALS in children. SMA affects roughly 1 in 6,000 live births, and 1 in 50 people are carriers of the disease. Although it’s very prevalent, we don’t hear much about the disease because most patients die before three years old, and there’s no publicity campaign like the ice bucket challenge for it,” Ma said. “Current therapies can only help a subset of patients and for some treatments, such as gene therapy, there’s already evidence that it may have strong side effects in the long-term. What causes the motor neuron degeneration in the pathogenesis of SMA has long been a question my lab is interested in.”
In the study, Ma and his team unexpectedly found the hyperactivation of cyclin-dependent kinase 5 (Cdk5), a kinase expressed in central nervous system neurons that causes mitochondrial defects and motor neuron degeneration.
First, investigators observed induced pluripotent stem cells from human SMA patients and SMA mice, and found Cdk5 activity was increased, according to the study.
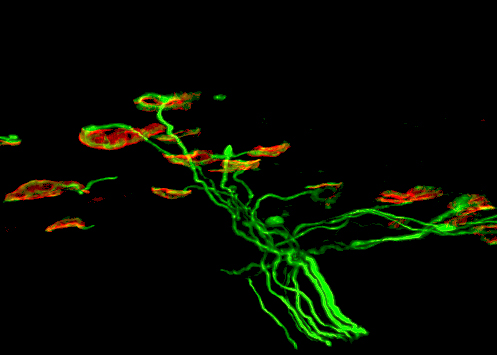
The scientists then reduced the activity of Cdk5 by genetically knocking out a Cdk5 activator in mice and by utilizing a pharmacological inhibitor for the human cells. Both methods produced a marked decrease in motor neuron degeneration and alleviated some of the main symptoms of SMA disease, including motor neuron hyperexcitability, excitatory synapse loss on motor neurons, and the denervation of motor neuron axons on muscles at neural muscular junctions.
The findings further scientists’ understanding of SMA and point to Cdk5 as a future direction for therapy development, Ma said. This study also revealed, for the first time, a positive feedback loop between Cdk5 and mitochondrial defects in maintaining calcium homeostasis. This feedback loop eventually leads to motor neuron excitotoxicity and degeneration, Ma said.
“In this study, we’ve identified an unexpected role of Cdk5 signaling in causing mitochondrial defects and selective motor neuron degeneration in SMA,” Ma said. “Based on our genetic rescue in both mouse models and in human induced pluripotent stem cell disease models, it really suggests a novel pathogenic mechanism and new therapeutic strategy for SMA. This could also have implications for other motor neuron diseases, such as ALS.”
Because currently available Cdk5 inhibitors come with high toxicity risks and low specificity, Ma said he hopes to develop a bioactive inhibitor which can target the pathway specifically to reduce the degeneration of motor neurons.
“Because our work also linked Cdk5 hyperactivation with mitochondrial defects, we’re also exploring how we could restore normal activity one step lower on the Cdk5 signaling cascade,” Ma said. “If we could target or restore the mitochondrial defects, that may help patients with SMA or ALS, and would be a completely new direction for treatment.”
The study was supported by National Institutes of Health grants R01NS094564, R21NS106307, and RF1AG077451, grants the Hartwell Foundation, Cure SMA, and the Agape Foundation.