The damaging effects of toxic proteins created in one inherited form of amyotrophic lateral sclerosis (ALS) are mediated by a protein called SPOP, according to a study published in the Proceedings of the National Academy of the Sciences (PNAS).
Reducing the abundance of SPOP or inhibiting its activity with a small molecule could protect neurons against protein toxicity, according to Robert Kalb, MD, director of the Les Turner ALS Center, the Joan and Paul Rubschlager Professor and co-senior author of the study.
“Several pharmacological agents are known to target SPOP and we are excited about the possibility of translating these observations into mice and potentially humans,” said Kalb, who is also chief of Neuromuscular Disease in the Ken and Ruth Davee Department of Neurology.
ALS is a progressive neurological disease that attacks motor neurons in the brain. There is no cure and life expectancy is typically three to five years from the onset of symptoms.
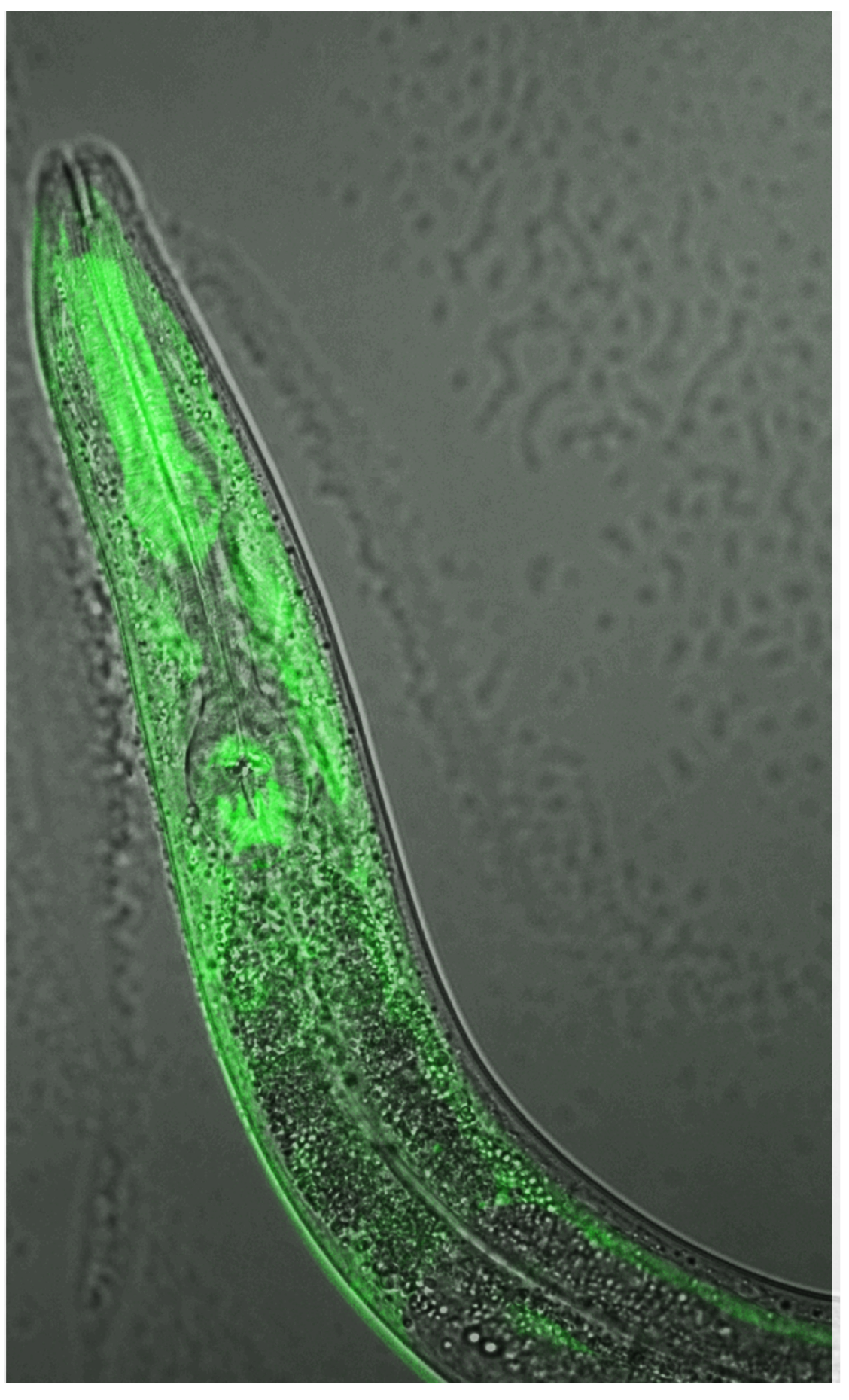
Most cases are sporadic, but some cases are caused by identifiable inherited mutations. The most common inherited cause is a mutation in a gene called C9ORF72 and this can lead to the production of highly toxic proteins.
In the current study, in collaboration with Todd Lamitina, PhD, associate professor of Cell Biology at the University of Pittsburgh, investigators studied the toxicity of dipeptide repeated proteins (DPR) PRx and GRx, both created by the mutation in C9ORF72.
Studying genetically modified Caenorhabditis. elegans (tiny worms that normally reside in soil) and vertebrate motor neurons — with and without the mutation — the scientists discovered that DPR toxicity depends on the ubiquitin ligase adaptor SPOP. Reducing the abundance of SPOP dramatically reduced motor neuron damage, according to the authors, and further study revealed a “druggable” pathway that could represent a therapeutic strategy.
“SPOP appears to be working by controlling the three-dimensional architecture of DNA in the nucleus of nerve cells,” Kalb said. “If targeting this pathway in patients with C9ORF72 mutations is beneficial, it may point to a broader role of SPOP in ALS.”
In addition, future work should investigate the role of SPOP in other forms of ALS, Kalb said.
“It will take more in-depth study to determine if the more prevalent sporadic ALS — that is, ALS not caused by mutations in C9ORF72 — will kneel to SPOP targeted therapy.”
This work was supported by the National Institutes of Health grants NS094921 and NS096319, NS05225 and NS087077, the Les Turner ALS Foundation and the Heather Koster Family Charitable Fund.