Northwestern Medicine scientists have discovered a new target for slowing treatment-resistant prostate cancer, according to a recent study published in Nature Communications.
Inhibiting an epigenetic regulator called DOT1L reduced growth of human prostate tumor cells while sparing healthy cells, said Sarki Abdulkadir, MD, PhD, the John T. Grayhack, MD, Professor of Urological Research, vice chair for research in the Department of Urology and senior author of the study.
“Only cells with androgen receptors were affected,” said Abdulkadir, who is also a professor of Pathology and a principal investigator of the Specialized Program of Research Excellence (SPORE) in prostate cancer at the Robert H. Lurie Comprehensive Cancer Center of Northwestern University. “Most prostate cancer arises in cells with the androgen receptor, so this could be a very good drug target.”
Prostate cancer is the most common cancer in adult men, but also among the most lethal when advanced. Prostate cells are reliant on the hormone androgen for growth and function.
Most prostate cancers are driven by an androgen receptor that can cause downstream upregulation in cell proliferation proteins, such as MYC. This growth can sometimes be kept under control with treatments that reduce androgen, but many cancers have mechanisms that essentially produce their own androgen, eventually rendering such therapies as ineffective.
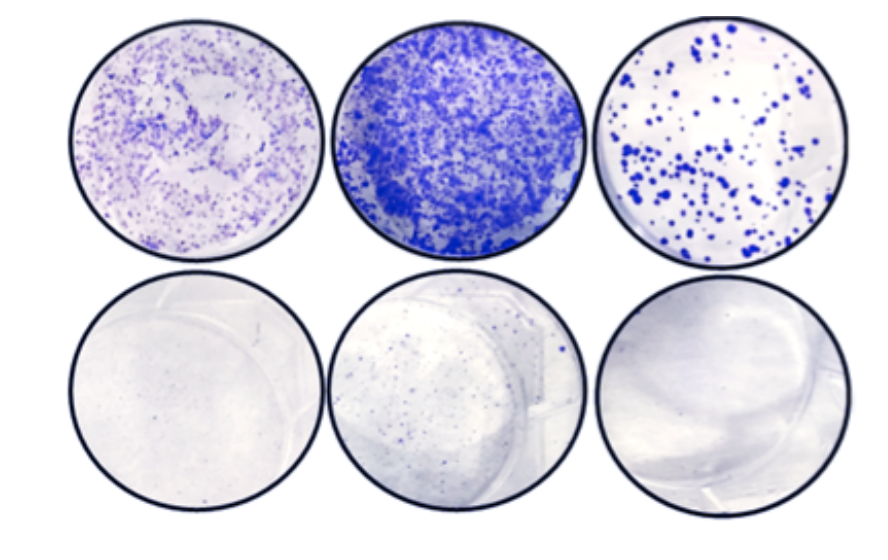
In the current study, Abdulkadir and his colleagues found DOT1L was upregulated in patients with prostate cancer across several large genetic datasets. Treating human prostate cancer cells with a DOT1L inhibitor suppressed the MYC pathway and blunted growth of cancer, but the mechanisms behind this were unclear, Abdulkadir said.
To learn more, the scientists conducted genome transcription and binding analysis to examine what happens when DOT1L is inhibited. They found the region of the genome that codes for MYC also contains sites regulated by both DOT1L and androgen receptors. In turn, MYC represses enzymes that degrade androgen receptors and MYC itself. Thus inhibiting DOT1L triggers a vicious cycle that further decreases MYC and androgen receptor levels, according to Abdulkadir.
“This negative feedback loop is part of why we saw such a dramatic effect,” Abdulkadir said.
However, the magnitude of the effect was not the most striking finding. That distinction belongs to the self-targeting nature of DOT1L inhibition. According to Abdulkadir, cells without androgen receptors don’t have the unique co-location of DOT1L, androgen receptors and MYC — meaning a DOT1L inhibitor may specifically target prostate cancer cells while leaving other tissues alone.
“DOT1L doesn’t exist just to give people cancer, it has a normal function throughout the body and if you inhibit it, it could cause side effects,” Abdulkadir said. “But some tissues, such as these androgen receptor positive cells, regulate DOT1L and MYC in a unique way. You could possibly use a smaller dose so other tissues are not dramatically affected.”
Abdulkadir and his collaborators have already begun searching for drugs that can inhibit DOT1L in humans, as the inhibitors currently available are not suitable for widespread therapeutic use.
“We are trying to identify compounds that can be used to block DOT1L because this could be really good for patients with treatment-resistant prostate cancer,” Abdulkadir said.
Rajita Vatapalli, PhD, recent graduate from the Driskill Graduate Program in Life Sciences (DGP) was lead author of the study. Debabrata Chakravarti, PhD, vice chair for Translational Research in the Department of Obstetrics and Gynecology, professor and associate director in the Division of Reproductive Science in Medicine and Pharmacology, and assistant director of shared resources at the Robert H. Lurie Comprehensive Cancer Center, was a co-author of the study.
Jindan Yu, MD, PhD, professor of Medicine in the Division of Hematology and Oncology and of Biochemistry and Molecular Genetics, was also a co-author of the study.
This work was supported by grants RO1CA123484, RO1CA196270, and P50CA180995 from the National Cancer Institute; and by the Department of Defense Prostate Cancer Research Program Awards W81XWH-14-2-0182, W81XWH-14-2-0183, W81XWH-14-2-0185, W81XWH-14-2-0186, and W81XWH-15-2-0062 to the Prostate Cancer Biorepository Network.