Many cancers hijack a metabolic mechanism known as the mTOR pathway to fuel growth and proliferation, and inhibiting that pathway could slow tumor growth, according to a Northwestern Medicine study published in Molecular Cell.
Inhibiting production of a key material produced by the pathway used for protein synthesis could be an effective strategy to slow tumor growth, according to Issam Ben-Sahra, PhD, assistant professor of Biochemistry and Molecular Genetics and senior author of the study.
“In cancer cells, mTOR is always ‘on,’ stimulating biosynthesis for the tumor cell to grow and proliferate,” said Ben-Sahra, who is also a member of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University. “We think this inhibitor could be used in combination with other therapies to more strongly reduce tumor growth.”
The mTOR pathway, which regulates cell growth in coordination with nutrient availability, helps control metabolism throughout the body. The pathway acts as a sensor: If enough nutrients are available, the pathway activates and facilitates biosynthesis of proteins.
In most cancers, the mTOR pathway is continually activated, fueling abnormal growth and proliferation. The pathway is also upregulated in rare genetic diseases called lymphangioleiomyomatosis (LAM) and tuberous sclerosis complex (TSC), in which non-cancerous tumors grow in many parts of the body.
However, the specific effects of mTOR activation in cancer and tumor syndrome disorders remain largely understudied, Ben-Sahra said. In the current study, Feinberg scientists used mouse models of cancer and cancer cell lines to investigate how cancer takes advantage of mTOR.
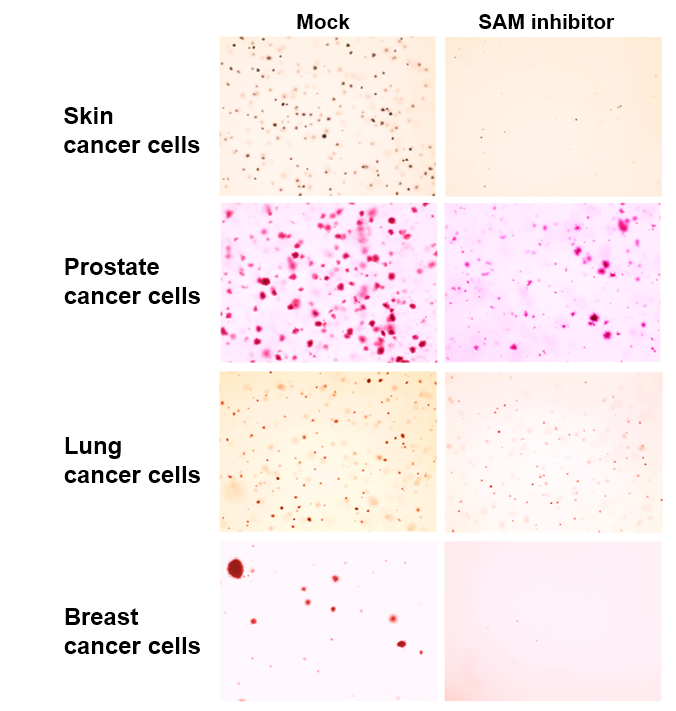
They found that different types of cancer — including breast cancer, prostate cancer and melanoma — have different mTOR regulation mechanisms, but all exhibited specific genes that activate mTOR.
“The cancers have different mutations that drive them, but they all have one thing in common: high mTOR signaling,” Ben-Sahra said.
This activation is different compared to normal cells, though. While mTOR is critical for all cell growth, cancer-driven mTOR activation specifically produces large amounts of S-adenosylmethionine (SAM), a metabolite required for protein synthesis. This presents an opportunity, Ben-Sahra said.
“We don’t want to target mTOR everywhere in the body; that could likely have some nasty side effects,” Ben-Sahra said. “But this new metabolic pathway is very important for cancer growth because it is highly activated.”

To investigate the phenomenon, Ben-Sahra and his collaborators created xenografts, transplanting patient-derived cancer cells into mice and applying the SAM inhibitor to these subjects. Though the inhibitor was originally designed as a tool for biochemical research, it had the effect of slowing cancer growth without serious consequences, according to Ben-Sahra.
“We didn’t know what to expect, but we were happy to see that all those proliferating cancer cells were affected but the mice did not show any obvious side effects,” Ben-Sahra said.
Ben-Sahra said he hoped that this tool could be refined for therapeutic use in the future, making it more effective and more specific to cancer cells and TSC cells.
“We think we can exploit this tool for cancer patients, using it as an adjuvant therapy,” Ben-Sahra said.
Elodie Villa, PhD, a postdoctoral fellow in the Ben-Sahra laboratory, was lead author of the study and was supported by the Tuberous Sclerosis Alliance for this project.
Co-authors of the study include Benjamin Singer, ’07 MD, ’10 GME, assistant professor of Medicine in the Division of Pulmonary and Critical Care and of Biochemistry and Molecular Genetics, and Peng Gao, PhD, research assistant professor of Medicine in the Division of Hematology and Oncology and director of the Lurie Cancer Center Metabolomics Core Facility at Northwestern University.
Animal work was performed with the help of the Northwestern Developmental Therapeutics Core.
Singer and Gao are members of the Lurie Cancer Center.
This work was supported by the Developmental Therapeutics Core at Northwestern University and the Robert H. Lurie Comprehensive Cancer Center National Cancer Institute grant CA060553; the Northwestern proteomics core; from the National Institutes of Health grants R00CA194192-04, R01GM135587, K08HL128867 and R01HL149883; the LAM Foundation Career Development Award LAM0127C01-18; the Tuberous Sclerosis Alliance postdoctoral fellowship (SP0057487) and the Philippe Foundation and Servier Institute prizes.