For the first time, scientists have characterized how a mutation in the gene KCNQ2, associated with pediatric epilepsy, affects human neuron activity, according to a Northwestern Medicine study published in eLife.
The findings suggest early therapy could be helpful but further study is still required, according to Evangelos Kiskinis, PhD, assistant professor in the Ken and Ruth Davee Department of Neurology Division of Neuromuscular Disease and co-senior author of the study.

“If our hypothesis is correct, one would need to initiate treatment early,” said Kiskinis, who is also an assistant professor of Physiology, a New York Stem Cell Foundation Robertson Investigator and a member of the Simpson Querrey Institute. “Now, we’re thinking of ways to take this forward.”
KCNQ2 is a gene that is responsible for neuronal M-current, a potassium current that regulates potassium channels. Mutations in KCNQ2 are a leading cause of early-onset pediatric epilepsy.
The association between KCNQ2 and pediatric neurodevelopmental epilepsy was highlighted only 10 years ago by several groups, including one led by John Millichap, MD, ’10, ’11 GME, associate professor of Pediatrics in the Division of Neurology and Epilepsy and co-author of the study. Since then, more and more cases have been found as clinicians learned about the condition and the variant was added to standard genetic screening panels.
While the symptoms had been catalogued in children, and mouse studies had revealed some aspects of disease, no one had examined these mutations in human neurons, according to Alfred George, Jr., MD, chair and Magerstadt Professor of Pharmacology and co-senior author of the study.
“Investigating the functional and physiological consequences of KCNQ2 mutations in a human neuron can provide greater insight into the molecular basis for this monogenic epilepsy,” said George, who also director of the Channelopathy-associated Epilepsy Research Center.
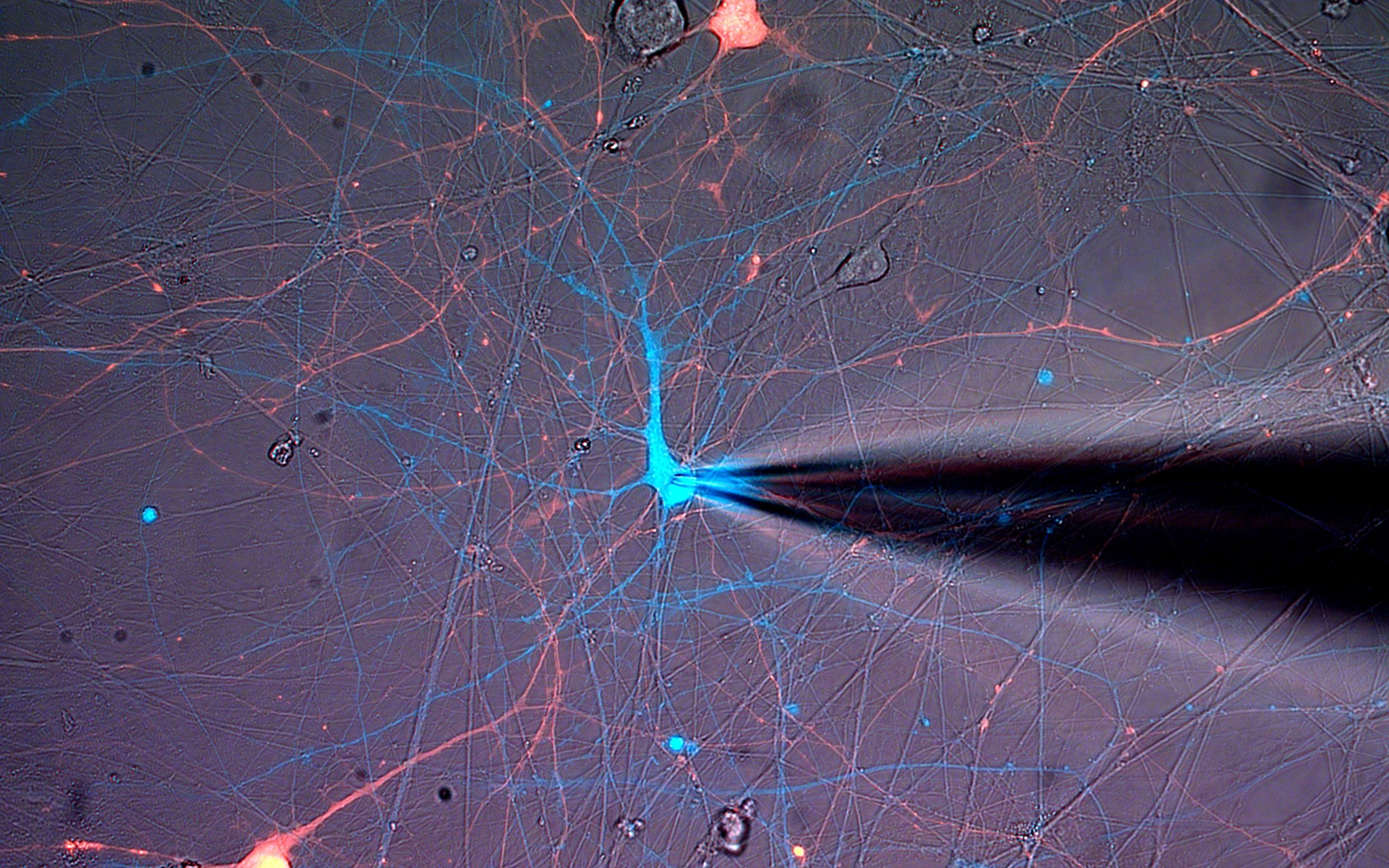
In the study, scientists isolated cells from patients with KCNQ2 epilepsy and used those cells to create model neurons with or without the mutation. They cultured the neurons and recorded electrophysiological activity, finding that the neurons exhibited a burst-suppression activity model: periods of high-voltage electrical activity alternating with periods of little activity.
This was highly reminiscent of the patterns seen on electroencephalograms (EEGs) of patients with the mutation, according to Dina Simkin, PhD, research assistant professor of Neurology in the Division of Neuromuscular Disease and lead author of the study.
“It was definitely surprising and exciting to find such similar burst-suppression-like firing patterns in a dish,” Simkin said.
Further analysis revealed that while the M-current was less active, other potassium channels were upregulated. This is likely the neuron’s attempt to compensate for the defective M-current, Simkin said, but this homeostatic regulation may be doing more harm than good.
“The electrical phenotype in patient neurons was associated with maladaptive compensation, rather than the loss-of-function KCNQ2 channel mutation,” Simkin said.

Thankfully, this problem could have a solution: A recently-developed small molecule drug can boost activity in the M-current. If this drug could be administered early in the course of disease, children with this mutation might be able to avoid the secondary channel upregulation.
“It would only be relevant early on, because beyond a particular time point, the last thing you need is more potassium current activation,” Kiskinis said. “On the other hand, our findings offer alternative targets to treat this disorder.”
Now, Kiskinis and his collaborators are testing this hypothesis while also learning more about the heterogeneity of the disease, exploring improved mouse models and creating more patient-derived neurons to examine broader relevance of their findings in the disease.
“We are curious if perhaps different genetic variants might lead to a different disease presentation through alternative pathways,” Kiskinis said. “So we’re interested in seeing if that is true, or whether this model that we came up with is broadly relevant for other patients.”
Other co-authors of the study include Steven Lubbe, PhD, assistant professor of Neurology in the Division of Movement Disorders; Peter Penzes, PhD, the Ruth and Evelyn Dunbar Professor of Psychiatry and Behavioral Sciences, professor of Physiology and Pharmacology, and director of the Center for Autism and Neurodevelopment; and Marc Forrest, PhD, research assistant professor of Physiology.
This study was supported by National Institutes of Health National Institute on Neurological Disorders and Stroke grant U54NS10887 and the New York Stem Cell Foundation.