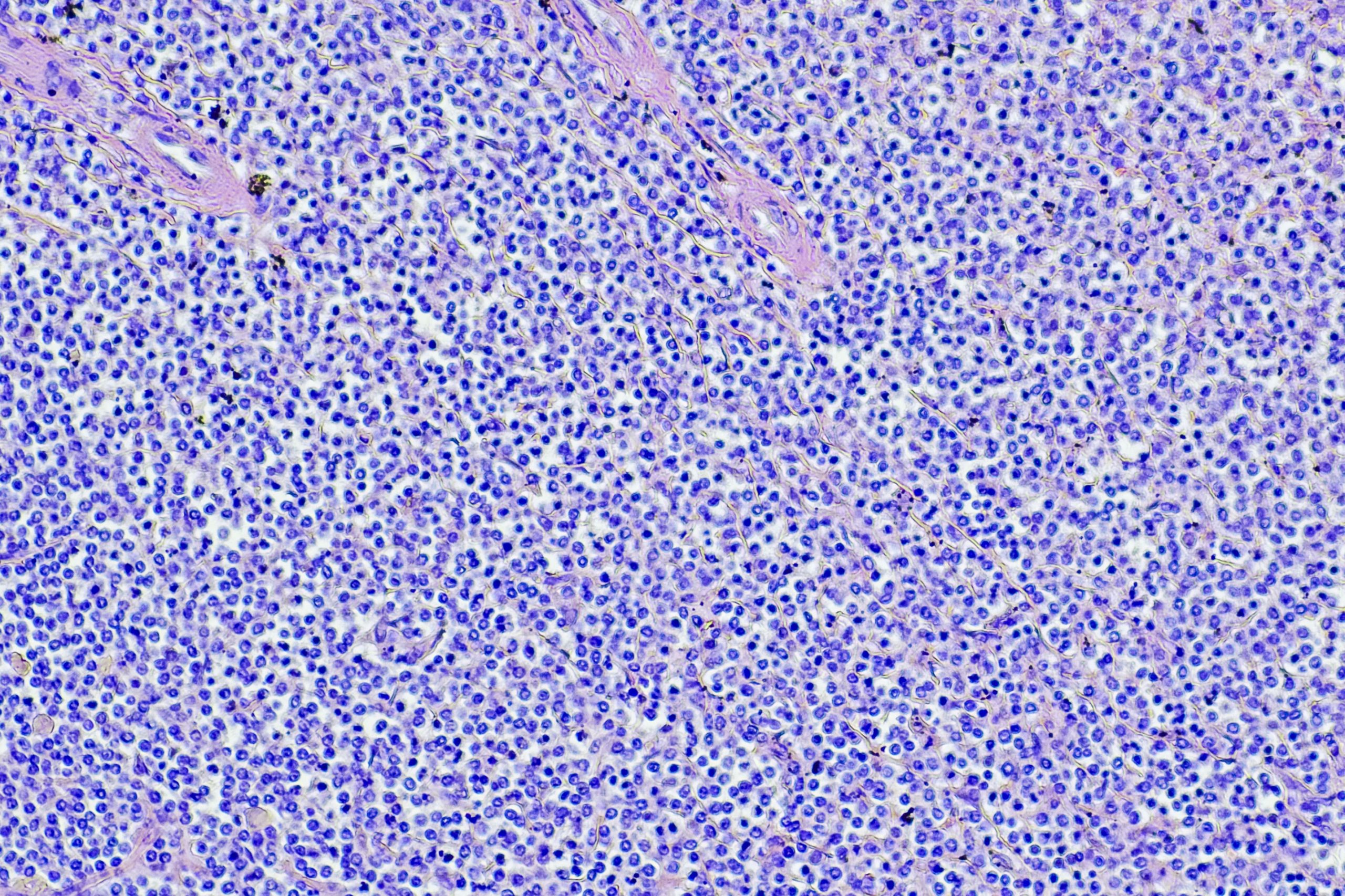
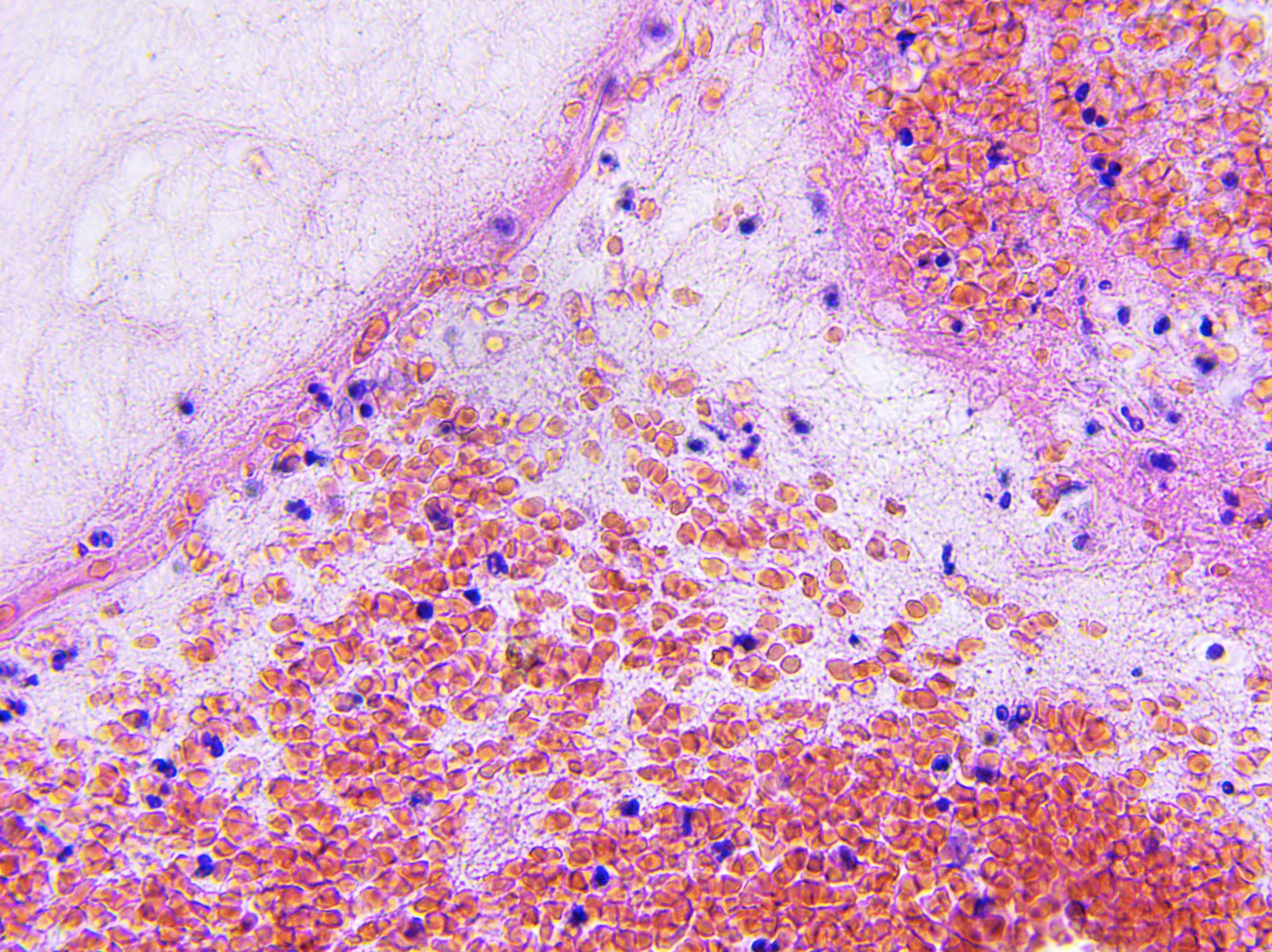
Defective cilia in the brain may play a role in some forms of severe schizophrenia, according to a study published in Nature Communications.
Deleterious mutations in PCM1, a gene that is necessary for the function of neuronal cilia, were linked with symptoms similar to schizophrenia in a variety of experimental animal models. Similar mutations were also observed in a cohort of human subjects with treatment-resistant schizophrenia, bolstering the theory that mutations in PCM1 — or another genetic issue that leads to shortened cilia — contribute to schizophrenia, according to Nicholas Katsanis, PhD, associate chief research officer for translational research at the Stanley Manne Children’s Research Institute at Ann & Robert H. Lurie Children’s Hospital of Chicago and senior author of the study.
“I cannot definitively say if it is the worsening cilia length that is causing this schizophrenic behavior, or if that is one part of the phenotype of these mutations,” said Katsanis, who also holds the Valerie and George D. Kennedy Research Professorship in Human Molecular Genetics and is a professor of Pediatrics. “But, this could explain why some patients are resistant to treatment.”
Cilia are small hair-like structures found on the surface of many cells. They are critical for embryonic development, and many genetic defects in cilia simply halt development altogether.
Cilia are present throughout the brain on the surface of neurons, and several genetic studies have hinted at links between cilia mutations and psychiatric illness. But, according to Katsanis, the precise role of cilia in the brain is unclear, especially after development.
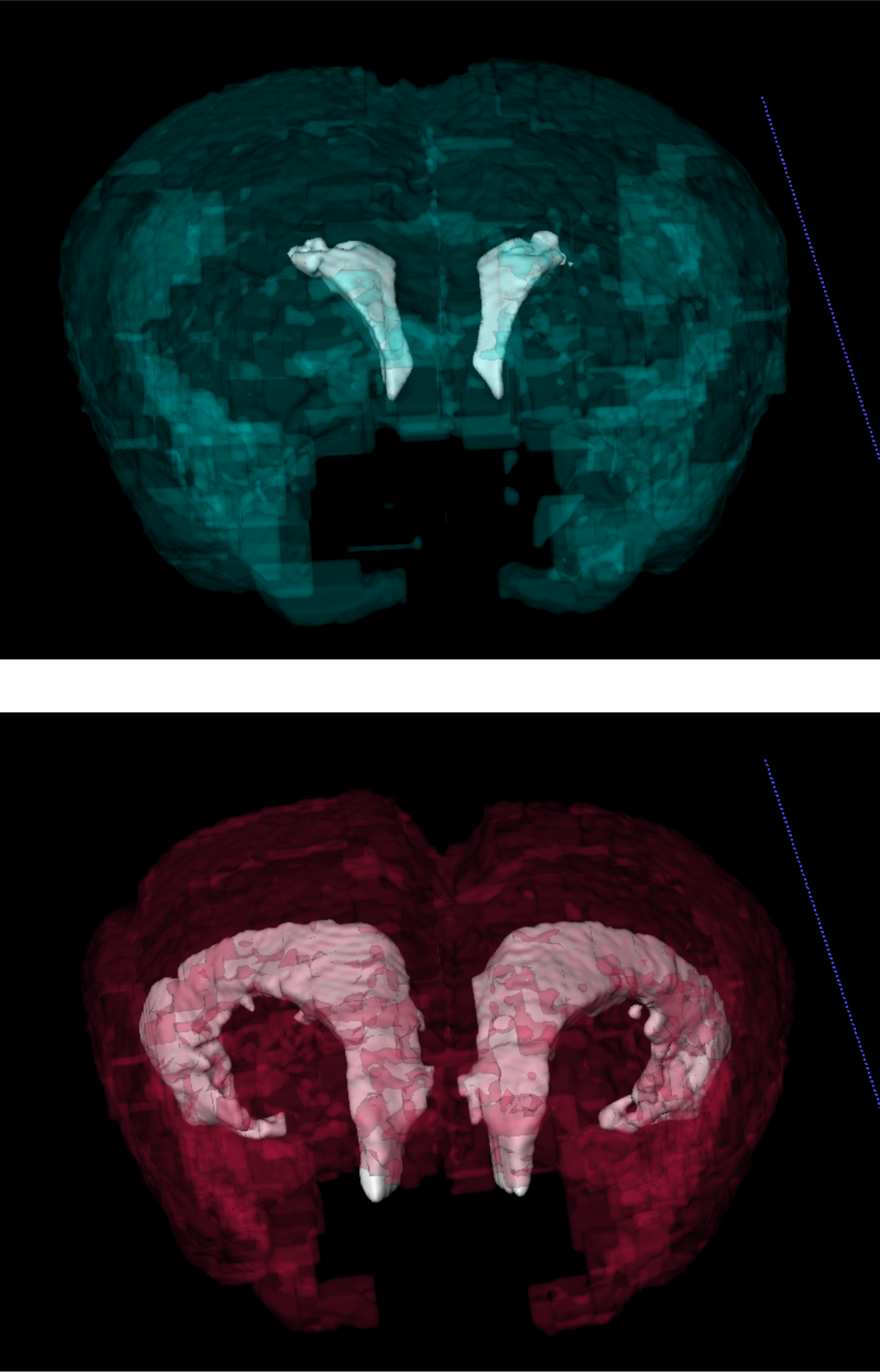
In the current study, the team studied PCM1, a gene that codes for a protein vital to cilia function. Previous studies had shown that when PCM1 is deleted in cell culture systems, cells fail to form cilia and often die. But when the investigators created mice that lacked PCM1, they were surprised to find them healthy at birth.
When the animals reached adolescence, however, they began exhibiting behavioral issues. At the same time, shorter-than-normal cilia started appearing in several (but not all) regions of the mouse brain.
“This was odd and unexpected,” Katsanis said. “But this observation tracks well with the timeline of schizophrenia development in people, where it is often first diagnosed during puberty.”
Shifting their focus to human subjects, the investigators assembled a cohort of patients who had severe schizophrenia, defined as people who failed to respond to potent antipsychotic drugs such as clozapine. They sequenced several cilia-related genes and found an excess of individuals with rare mutations in PCM1 compared to unaffected people.
Katsanis and his colleagues then took those mutations and inserted them into zebrafish, finding that, as predicted, they interfered with gene function. In search of mechanistic clues, they returned to the mouse model. There, they found a defect in the expression of genes that regulate dopamine receptors in neurons — the same receptors targeted by clozapine.
“Our working model is that in the absence of PCM1, fewer dopamine receptors rise to the surface of the cell,” Katsanis said. “If you have such a deficit, your ability to respond to dopamine drugs is reduced.”
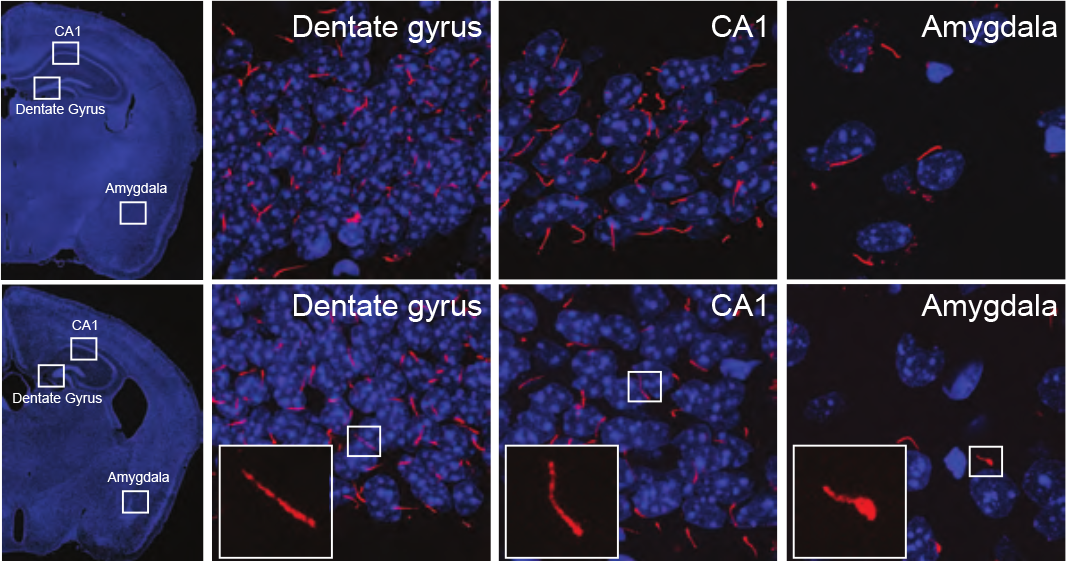
While Katsanis is careful to acknowledge that more work is required before one could claim shortened cilia cause schizophrenia, these findings do intimate a mechanistic link between the genetic finding and the excess PCM1 involvement in a subset of people with schizophrenia. This conclusion is only possible due to the breadth of experimental techniques utilized in the study, according to Tanner Monroe, PhD, postdoctoral fellow in the Katsanis laboratory and lead author of the study.
“The signal we detected by statistical methods alone in the patient population would not be overly compelling,” Monroe said. “It took the combination of in vivo modeling of the alleles in zebrafish to reveal a robust signal, followed by mechanistic insights from the rodent model to really bolster our confidence about the PCM1-SZ association.”
Beyond the specifics of PCM1, Katsanis believes that this study is a prime example of heterogeneity in psychiatric disorders — and how relying on genome-wide association alone could cause scientists to miss important clues and new pathways.
“Most scientists would agree that schizophrenia is not a single disease, but rather the manifestations of hundreds of diseases put together,” Katsanis said. “In that respect, large genome-wide sweeps help us discover common truths about the end disease, but not about its subtypes. So, although genome-wide analyses are a great step, we must now dig deeper.”
“For one, we ought to stratify individuals based on specific clinical characteristics,” Katsanis continued. “For another, we must come up with ways to synthesize different sources of data. A single approach, genetic or biological, is not going to provide sufficient resolution to understand this disorder.”
This work was supported by National Institutes of Health grants 5T32DK108738-04 and U54 HG002373 and the Silvo O. Conte Center grant MH084018.