Northwestern Medicine scientists have identified how genetic mutations associated with many cancers drive proliferation, according to a study published in Molecular Cell.
Mutations in the genes RAS and RAF allow cancer cells to create their own nucleotides, one of the molecular building blocks of cells throughout the body. While this helps cancers spread, it also represents an opportunity for intervention, according to Issam Ben-Sahra, PhD, assistant professor of Biochemistry and Molecular Genetics and senior author of the study.
“Now that we’ve identified this molecular connection, we can imagine targeting this pathway,” said Ben-Sahra who is also a member of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University. “This could be one of the achilles’ heel of the cancer.”
All cells in the body require molecules such as lipids, proteins and nucleotides to create macromolecules to maintain cell function. However, most normally growing cells in the body don’t divide all the time. Instead, they require specific growth signals to activate their cellular metabolism, such as in case of injury, infectious diseases and other complicating factors.
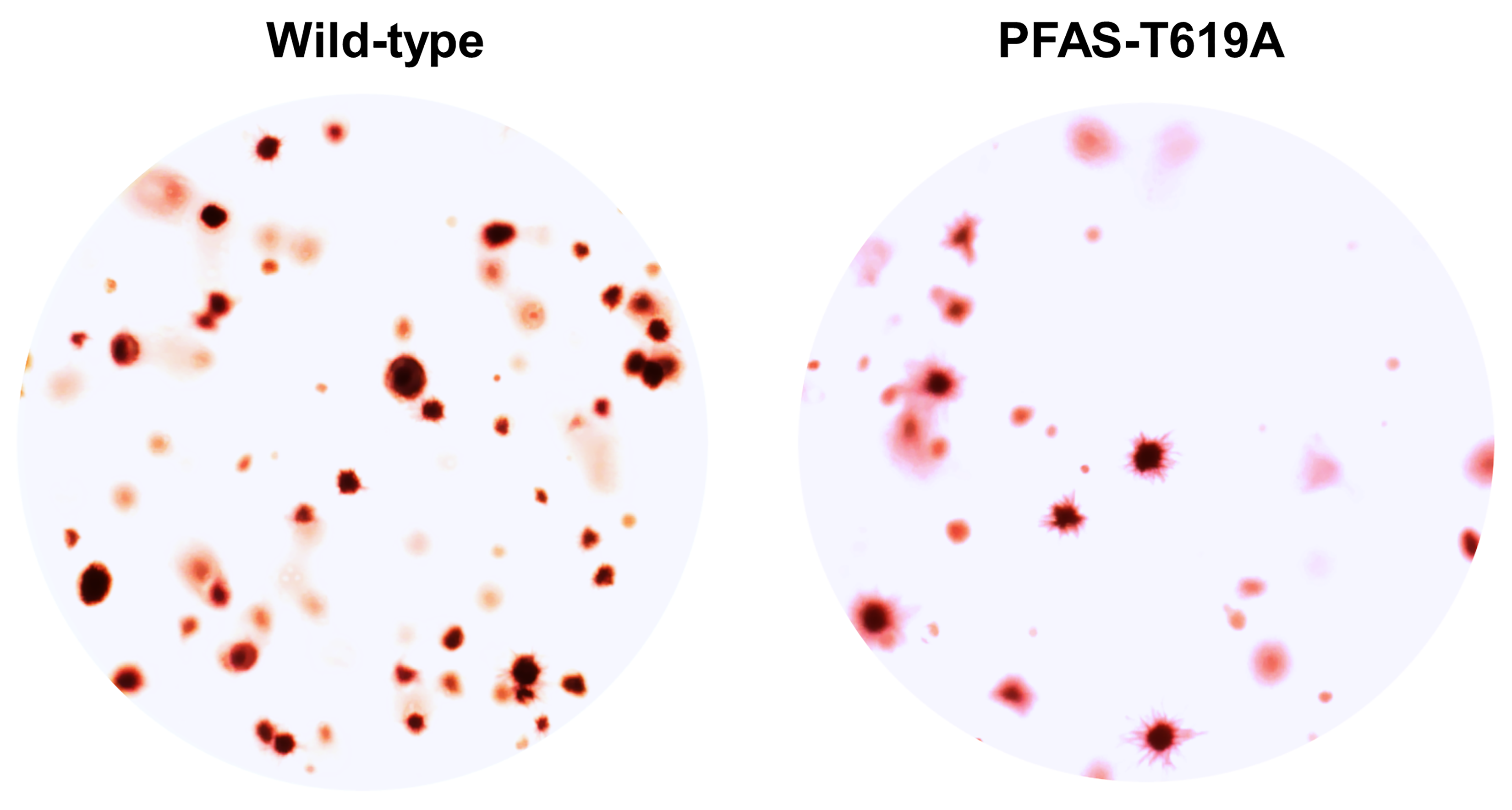
Cancer cells, on the other hand, grow continually, which requires them to make their own macromolecules without any input from growth signals. Some scientists believed that in some cancers, RAS and RAF mutations might be the culprit, according to Ben-Sahra.
“We wanted to understand how cancer cells hijack their metabolism to proliferate,” Ben-Sahra said.
Cancer-causing mutations in the genes RAS and RAF have been known to affect cellular metabolism, but exactly how they helped trigger cell growth was unclear. To investigate, Ben-Sahra — along with co-first authors Eunus Ali, PhD and Umakant Sahu, PhD, both postdoctoral fellows in the Ben-Sahra laboratory — analyzed cells with the RAS and RAF mutations. They found that through the activation of the target ERK, cancer cells can directly and rapidly stimulate nucleotide synthesis.

Working with John Asara, PhD, at the Beth Israel Deaconess Medical Center and Peng Gao, PhD, research assistant professor of Medicine in the Division of Hematology and Oncology and director of the Lurie Cancer Center Metabolomics Core Facility at Northwestern University, the investigators tracked the newly synthesized nucleotides using metabolomics technology. They found that RAS and RAF-mutated cells had greater numbers of nucleotides created from activation of the RAS-RAF-ERK pathway when compared to normal cells.
“With the RAS and RAF mutations, cancer cells can rapidly reprogram their metabolism and grow without any physiological signals,” Ben-Sahra said. “That’s what makes them cancerous.”
Previous attempts to therapeutically target cancer cell proliferation have used RAF inhibitors, but these experimental treatments often did not work long-term, according to Ben-Sahra.

“It might work at the beginning, but by the end of the treatment the patients would often exhibit resistance,” Ben-Sahra said.
Instead, targeting both RAS and RAF and the regulated nucleotide metabolism may represent a more promising therapy, one that will effectively cut off homegrown nucleotides whilst being less vulnerable to treatment resistance.
“We’re hoping it could have some synergistic effect,” Ben-Sahra said.
This study was supported by the Lynn Sage and LAM Foundations grant LAM0127C01-18 and National Institutes of Health grants R00CA194192, R01GM135587, 5P01CA120964 and 5P30CA006516.