A new Northwestern Medicine study has uncovered a signaling process that regulates inflammation, findings that may help scientists block the immune system’s white blood cells from causing the out-of-control inflammation that can damage organs in many autoimmune diseases.
In a paper published in the Journal of Experimental Medicine, William Muller, MD, PhD, Magerstadt Professor and chair of Pathology, and his lab focused on a molecule called CD99, a key component of transendothelial migration (TEM), the process of white blood cells – known as leucocytes – leaving the bloodstream and entering tissues.
Dr. Muller has been studying CD99 for more than 15 years; in 2002, he discovered that inhibiting its function with antibodies also stops inflammation. But up until now, scientists didn’t know how the molecule regulated TEM.
“The problem was that CD99 is a unique molecule; essentially unlike any other encoded by the human genome,” Dr. Muller said. “There were no clues that could be gained by studying its sequence or structure to tell us how it transmitted a signal inside the endothelial cell to control this process.”
So first author Richard Watson, an MSTP student, designed an elegant series of experiments to find out how CD99 transmits its signal. The team started by using an anti-CD99 antibody to interrupt the signaling that occurs when CD99 on leucocytes binds to CD99 on the endothelial cells that line blood vessels, allowing the leucocytes to pass into affected tissues.
“Basically, we reasoned that if we activated the signaling pathway downstream of CD99, we could overcome the block to leukocyte emigration that was mediated by the anti-CD99 antibody,” Dr. Muller said. “Richard identified several agents that could overcome the anti-CD99 block. The common denominator was that they could all activate protein kinase A (PKA).”
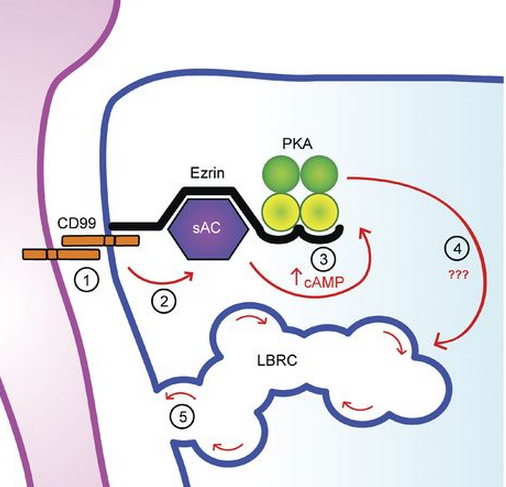
They worked backwards from there to fill in the chain of events that connects CD99 and transmigration: They found that a second messenger, cyclic adenosine monophosphate (cAMP), activated PKA, and that a unique enzyme, soluble adenylyl cyclase (sAC), produced the cAMP (see diagram). The scientists went on to show in vitro and in vivo that blocking sAC with a known small molecule inhibitor stopped inflammation. They witnessed the same effect in mouse models missing sAC in their endothelial cells.
“Understanding how CD99 functions provides us with more opportunities to control it,” Dr. Muller said. “Now that we know how CD99 works inside the endothelial cell at the molecular level, we can begin to target these other molecules and their interactions with CD99. For example, one of the key molecules in the pathway is a unique soluble adenylyl cyclase for which a small molecule inhibitor already exists. We show in the paper that this drug blocks the function of CD99 in a mouse model of dermatitis and greatly inhibits inflammation.”
In the future, the study’s findings could also be applied to augment inflammation in patients that are immunodeficient.
“It’s important to study the basic principles behind the body’s regulatory processes,” Dr. Muller said. “Someday we might want to develop a drug that turns these pieces on, not just one that turns them off.”
This study was supported by National Institutes of Health grants F30 HL116100-01A1, R37 HL064774, R01 HL046849, R01 GM62328 and R01 GM107442 and the Sidney Bess Eisenberg Memorial Fund.