A recent Northwestern Medicine study has discovered a previously unknown molecular mechanism that supports antitumor responses and cell survival in cytotoxic immune cells, according to findings published in the Journal of Clinical Investigation.
CD8+ T-cells are cytotoxic immune cells that can kill cancer cells, as well as cells infected with viruses or bacteria. These specialized T-cell express receptors that recognize specific antigens and stimulate an immune response to attack cancer cells and other foreign antigens.
Adoptive cell therapy, a type of immunotherapy that includes CAR T-cell therapy and tumor-infiltrating lymphocyte therapy, uses the patient’s own T-cells to fight cancer and other diseases.
However, this persistent immune response causes T-cells to become exhausted and lose their ability to kills cancer cells. Increasing T-cell survival especially within the tumor microenvironment, has remained an ongoing area of interest in the field, according to Bin Zhang, MD, PhD, the Johanna Dobe Professor of Cancer Immunology and senior author of the study.
“In this study, we tried to think about how use metabolic manipulation to make effector T-cells persist longer, which increases T-cell survival, while actually still maintaining their effector function which means they still have ability to kill tumor cells,” said Zhang, who is also a professor of Medicine in the Division of Hematology and Oncology, of Microbiology-Immunology and of Pathology.
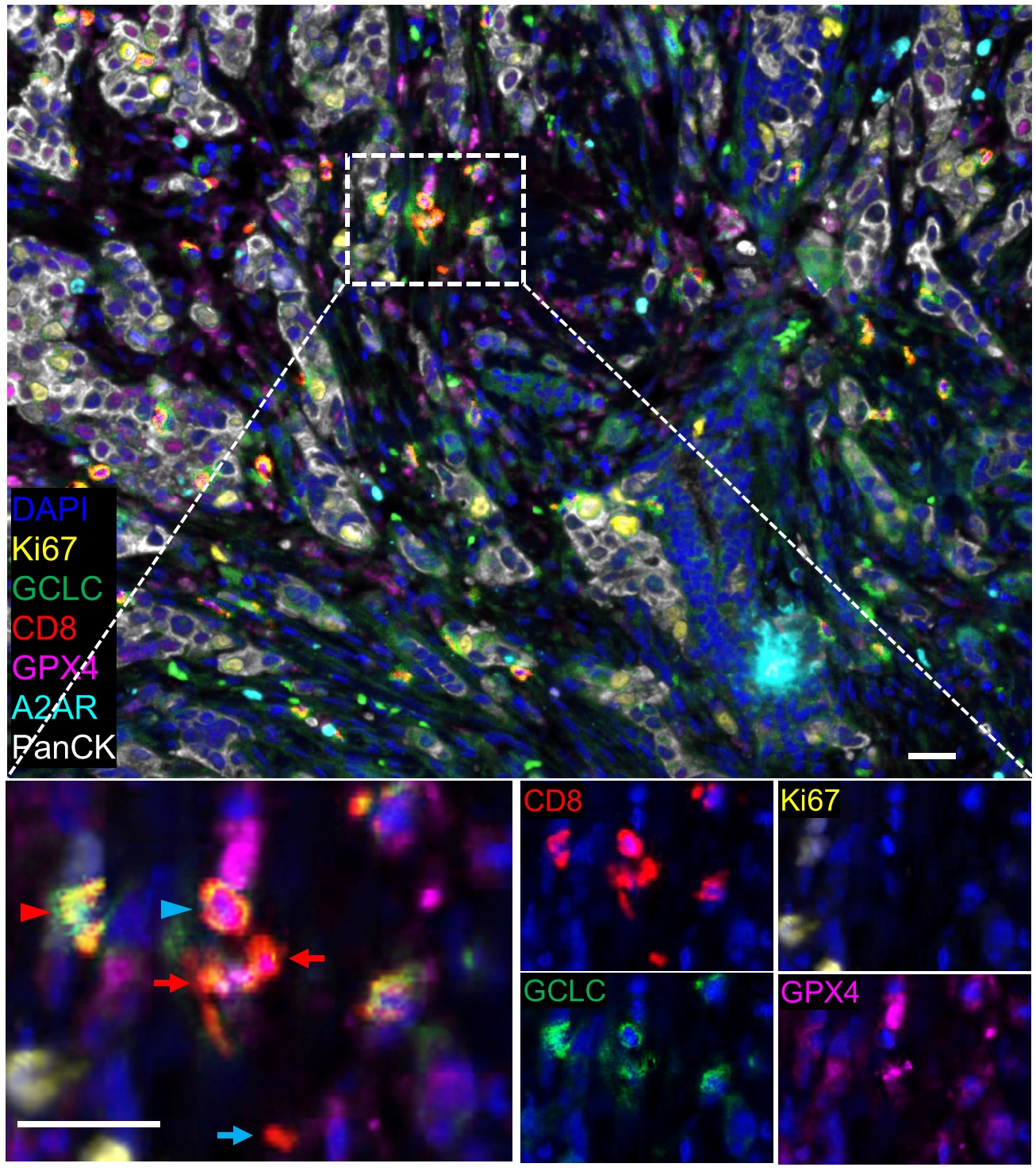
Previous work has shown that increased levels of a metabolite called glutathione (GSH), a basic nutrient that supports cell survival, are expressed in many tumors and support cancer progression and treatment resistance. Antitumor responses and proper CD8 T-cell function also relies on the enzyme glutathione peroxidase 4 (GPX4), and increased GSH has been shown to prevent GPX4-dependent ferroptosis, a type of programmed cell death.
In the current study, Zhang and his team aimed to identify the molecular mechanism in the GSH-GPX4 metabolic axis that modulates the antitumor response of CD8 T-cells.
By selectively modulating CD8+ T-cells with pharmacological blockade and genetic ablation techniques, and using RNA-sequencing and metabolic profiling, the investigators found that the adenosine A2A receptor (A2AR) signaling pathway coordinates with the GSH-GPX4 axis to support the metabolic fitness and antitumor function of CD8+ T-cells.
“We identified that this signaling pathway [A2AR] closely crosstalks with one of the essential metabolic pathways, GSH-GPX4, which is key for maintaining T-cell survival,” Zhang said.
Subsequent analysis of single-cell sequencing data also revealed a subtype of intra-tumoral CD8+ T-cells that expressed a GSH metabolism-related gene signature associated with therapeutic response and survival across several types of cancers, including melanoma, breast cancer and colon cancer.
According to Zhang, the findings could inform the development of new immunotherapeutic strategies that support increased T-cell activation and survival within the tumor microenvironment, as well as support the discovery of new biomarkers that predict immunotherapy response.
“We propose a mechanism underlying an antitumor response of CD8+ T-cells by modulating GSH metabolism and the A2AR signaling,” Zhang said.
Co-authors include Jason Miska, PhD, assistant professor of Neurological Surgery; Jennifer Wu, PhD, the Mary and Patrick Scanlan Professor of Urology and Microbiology-Immunology; Deyu Fang, PhD, the Hosmer Allen Johnson Professor of Pathology; Jeffrey Sosman, MD, professor of Medicine in the Division of Hematology and Oncology; and Navdeep Chandel, PhD, the David W. Cugell, MD, Professor of Medicine in the Division of Pulmonary and Critical Care and of Biochemistry and Molecular Genetics.
Zhang, Miska, Wu, Fang, Sosman and Chandel are members of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University and the Center for Human Immunobiology.
This research was in part supported by the National Institutes of Health grants CA222963, CA250101, 611 CBC Catalyst Award, CA257520 and CA258857.