Northwestern Medicine scientists have discovered that mitochondria are not necessary for the proliferation of specialized immune cells in the central nervous system, but do help those cells respond to demyelinating injuries, according to a recent study published in Nature Metabolism.
“Microglia we know are connected to many neurological diseases, including decline in cognition during normal aging and Alzheimer’s Disease. People think that mitochondria are related to all these diseases. So, we asked whether mitochondria and microglia are in any way playing a role and, to our surprise, largely no – other than in demyelinating diseases,” said Navdeep Chandel, PhD, the David W. Cugell, MD, Professor of Medicine in the Division of Pulmonary and Critical Care and of Biochemistry and Molecular Genetics and senior author of the study.
Microglia are tissue-resident immune cells that support proper brain development and maintenance. Microglial dysfunction is known to contribute to Alzheimer’s disease, suggesting that microglial function may be a possible therapeutic target for treating central nervous system diseases. Microglia also drive immune responses that promote recovery after demyelinating injury in the central nervous system.
Microglial function has also been linked to mitochondrial function, specifically the mitochondrial respiratory chain, a protein complex found within the inner mitochondrial membrane that the Chandel laboratory previously demonstrated supports other immune responses. Whether the mitochondrial respiratory chain is necessary for the function and proliferation of microglia, however, has remained unknown.
Previous work from Chandel’s laboratory, published in Nature, showed that cancer cells will not proliferate in the absence of mitochondrial complex III, one of five protein complexes in the mitochondrial inner membrane that create energy, or ATP, for the cell. This demonstrated that mitochondrial dysfunction impairs cancer growth.
In the current study, the investigators deleted the mitochondrial complex III subunit Uqcrfs1 in the microglia of adult mice to determine if the microglial respiratory chain is essential for the survival and proliferation of microglia. Surprisingly, they found it wasn’t.
“What we found, to our surprise, is in adult mice microglia that don’t have the mitochondrial complex III, they proliferate fine,” Chandel said. “So, it suggests that microglia are quite different, that they’re quite hearty bioenergetically and that they can live off glycolysis.”
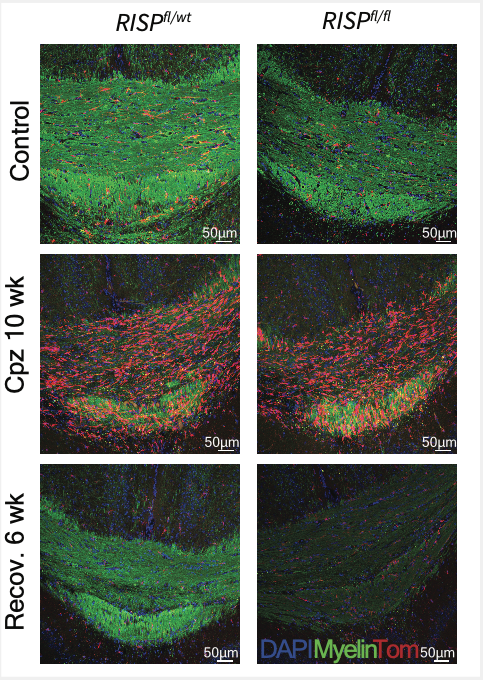
Subsequent behavioral analysis via the Barnes maze, a spatial learning and memory test revealed, that the loss of mitochondrial respiratory chain function was not enough to induce decline in spatial learning and memory in the adult mice.
“Mitochondrial dysfunction is not sufficient to give you an aged phenotype, this is probably one of the more surprising but interesting findings of the study” Chandel said.
Next, the investigators used mouse models of familial Alzheimer’s disease (5xFAD) to study amyloid-beta plaque deposition. It is thought that amyloid-beta plaques disrupt normal cell function in the brain and contribute to cognitive decline in Alzheimer’s disease.
The Chandel Lab team, surprisingly, found a decrease in hippocampal amyloid-beta plaque deposition, and increased interactions between the microglia and amyloid-beta plaques in mice with mitochondrial complex III-deficient microglia. However, they did not see any acceleration in cognitive decline in the 5xFAD mice that had mitochondrial complex III-deficient microglia.
“This indicated that mitochondrial respiratory chain in microglia was not required for protective responses to amyloid-beta plaque deposition,” Chandel said.
They did discover, however, that microglial mitochondrial respiratory chain function is required for recovery following a demyelinating injury. The scientists induced demyelination in the mice with a chow containing cuprizone, a copper chelating agent that causes demyelination patterns with similar characteristics to multiple sclerosis. When the cuprizone was removed, microglia were critical for remyelinating axons in the central nervous system to restore normal nerve function.

“That is massively impaired if you don’t have functional mitochondria in your microglia,” said Joshua Stoolman, PhD, a research associate in the Chandel laboratory and lead author of the study.
The findings suggest that microglial mitochondrial respiratory chain function is required for distinct microglial responses in the adult central nervous system, according to the authors.
Co-authors include Rogan Grant, PhD, a Schmidt Science Fellow at Northwestern; Samuel Weinberg, ‘19 MD, ‘19 PhD, assistant professor of Pathology in the Division of Experimental Pathology; Taylor Poor, MD, PhD, a clinical fellow in the Chandel laboratory; Karis D’Alessandro, a student in the Driskill Graduate Program in Life Sciences (DGP); Jerica Tan, a student in the Medical Scientist Training Program (MSTP); Karen Ridge, PhD, the Ernest S. Bazley Professor of Pulmonary Sciences; Paul Schumacker, PhD, professor of Pediatrics in the Division of Neonatology, of Cell and Developmental Biology and of Medicine in the Division of Pulmonary and Critical Care; and Scott Budinger, MD, chief of Pulmonary and Critical Care in the Department of Medicine and the Ernest S. Bazley Professor of Airway Diseases.
Chandel, Schumacker and Budinger are members of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University.
This work was supported by the National Institutes of Health grants 2P01AG049665-06, 2T32AI083216-11 to and 5T32HL076139-18.