Northwestern Medicine investigators led by Derek Walsh, PhD, professor of Microbiology-Immunology, have discovered how poxviruses disarm and evade mitochondrial-driven antiviral responses for their replication in host cells, according to findings published in Nature Communications.
Nathan Meade, PhD, a postdoctoral fellow and research associate in the Walsh Lab, was lead author of the study.
Poxviruses are a family of large, double-stranded DNA viruses, which include variola virus (smallpox), monkeypox virus, and others, that infect both humans and animals. This category of viruses is unusual in that they replicate entirely in the cytoplasm of host cells and form large DNA-filled replication compartments, which leave them vulnerable to detection by nucleic acid sensors, including one known as cGAS. In response, the virus encodes nearly 100 immunomodulator proteins in the early stages of infection to help it evade antiviral response pathways.
Poxviruses also encode a protein called F17, which is expressed in later stages of infection and is required to block antiviral responses, as previously discovered by the Walsh laboratory. The nature of these antiviral responses and how F17 targets them, however, have remained unknown.
Using immunofluorescence and metabolic assays, Walsh’s team studied human cell cultures infected with vaccinia virus, the virus used for smallpox vaccination, and discovered that F17 blocks a mitochondrial-driven antiviral response that involves hyperfusion in infected cells.
“Specifically, as poxviruses replicate within the cell, the replication stress induces mitochondria to undergo a process called hyperfusion,” Walsh said.
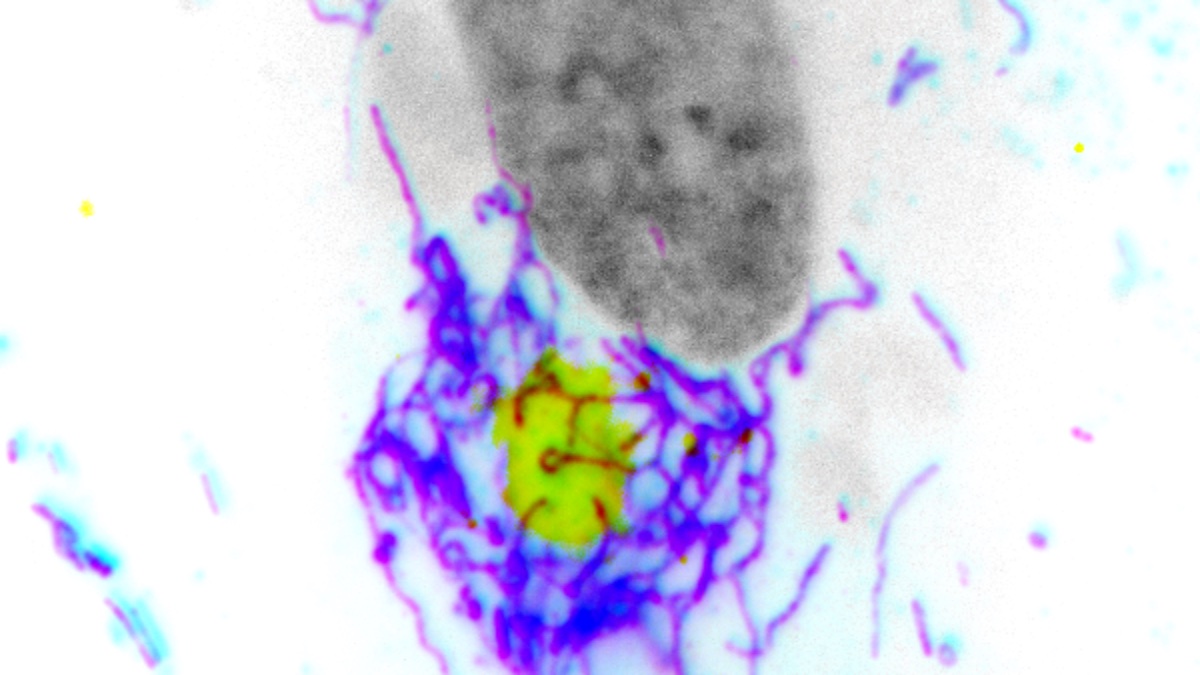
This hyperfusion process has two key functions: releasing mitochondrial DNA (mtDNA) to activate cGAS and driving cells to increase glycolysis — a metabolic pathway that converts glucose into energy for the cell — to support the production of antiviral proteins.
Instead, F17 counteracts these responses by localizing to mitochondria and dysregulating mTOR, a key metabolic sensor and kinase that controls both cGAS stability and glycolytic activity, according to Walsh.
“Overall, this highlighted how cytoplasmic viral DNA is not the only form of DNA that serves to alert cells to poxvirus infection, and that mtDNA and mitochondrially-controlled metabolic changes also play a key role in sensing and responding to the progression of poxvirus replication,” Walsh said.
According to Walsh, the findings could support the future development of a potential therapeutic approach that involves targeting F17 or altering metabolic processes in poxvirus-infected cells, which may enhance the host’s ability to fight off infection.
“Indeed, F17 is not expressed well by attenuated poxviruses that are used as vaccines, and this may be one of the reasons that these vaccines elicit such a strong response,” Walsh said.
Walsh said his team is now interested in identifying the mechanisms by which F17 affects host cell metabolism and on host antiviral responses.
Co-authors of the study include Ram Chakrabarty, a student in the Driskill Graduate Program in Life Sciences (DGP), and Navdeep Chandel, PhD, the David W. Cugell, MD, Professor of Medicine in the Division of Pulmonary and Critical Care and of Biochemistry and Molecular Genetics.
This work was supported by National Institutes of Health grants P01 HL154998, P01 AG049665, R01 AI127456, R01 AI179744, and a Northwestern University Pulmonary and Critical Care Cugell predoctoral fellowship.