A new Northwestern Medicine study has uncovered previously unidentified intracellular mechanisms in the peripheral nervous system that cause Charcot–Marie–Tooth Type 2B disease, a rare congenital disorder that causes sensory deficits and muscle atrophy and weakness.
The findings improve the understanding of the origins of the disease and may also inform the development of new targeted therapies, according to the study published in the Proceedings of the National Academy of Sciences.
“These findings are important as they highlight an essential role for properly regulated mitochondria-lysosome contact site dynamics and function in the axons of sensory peripheral neurons, and demonstrate that this may be an important pathway in the pathogenesis of Charcot-Marie-Tooth Type 2B disease,” said Yvette Wong, PhD, assistant professor of in the Department of Neurology’s Division of Movement Disorders and co-lead author of the study.
Charcot-Marie-Tooth Type 2 diseases are a group of hereditary neuropathic disorders characterized by the degeneration of axons in peripheral nerves.
Charcot–Marie–Tooth Type 2B disease is specifically caused by mutations in a GTPase protein called Rab7, which leads to the degeneration of axons of peripheral sensory neurons. Wong and other Northwestern Medicine investigators previously found that mitochondria-lysosome contact sites can form to support essential crosstalk between mitochondria and lysosomes, and that the untethering of these contact sites is driven by Rab7’s GTPase activity. In the context of Charcot–Marie–Tooth Type 2B disease, Wong and colleagues also previously discovered that disease mutant Rab7 prevents the untethering of these contact sites, resulting in downstream defects in mitochondrial dynamics.
In the current study, Wong and colleagues aimed to determine in peripheral sensory neurons whether mitochondria-lysosome contact sites disrupted by mutant Rab7 lead to mitochondrial defects and whether those defects preferentially occur in axons in peripheral neurons, as observed in patients with the disease, or in the neuronal cell body.
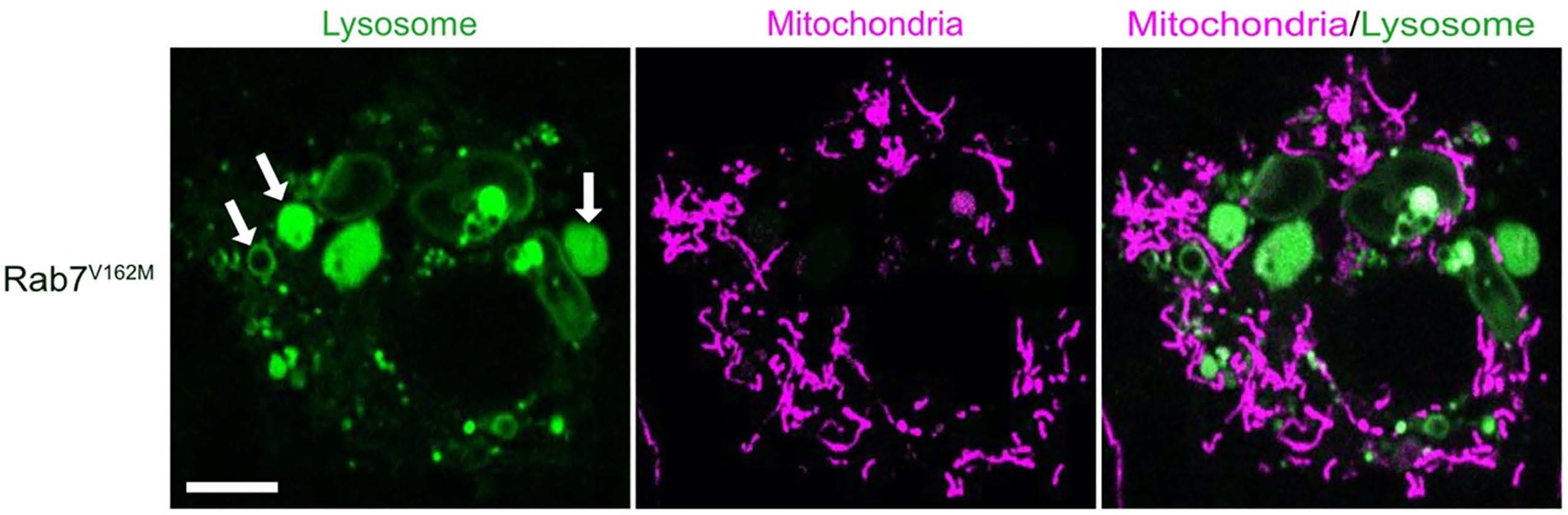
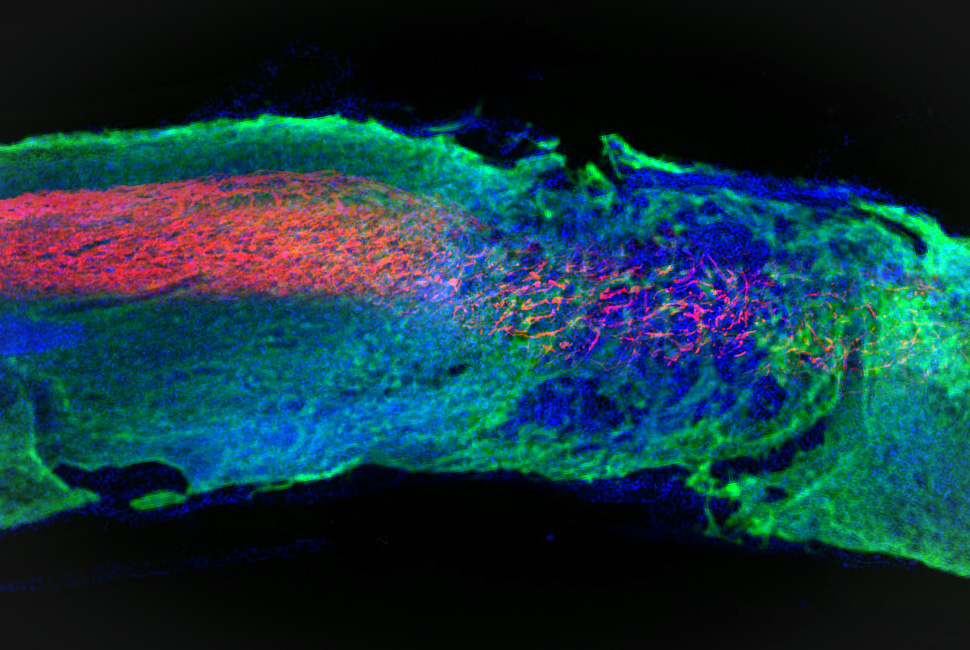
The investigators created a new mouse model of Charcot-Marie-Tooth Type 2B disease mutant Rab7; the mice displayed sensory behavior defects and neuropathy but had normal motor behavior. Using super-resolution and live microscopy to study the peripheral sensory neurons from these mice, the investigators identified mitochondria-lysosome contact sites that could not efficiently untether in axons. Importantly, promoting the untethering of mitochondria-lysosome contact sites was sufficient to improve mitochondrial dynamics in the axons.
The findings suggest that targeted therapies which help improve mitochondria-lysosome contact site tethering dynamics and function may improve mitochondrial health in these axons, according to the authors.
These findings also suggest that mitochondria-lysosome contact sites may play an important role in other genetic forms of Charcot-Marie-Tooth Type 2 disease, which also exhibit axonal degeneration, as well as in other neurological disorders that have mitochondrial dysfunction or axonal degeneration, according to Daniela Maria Menichella, MD, PhD, ‘08, ‘11 GME, associate professor of Neurology in the Division of Neuromuscular Disease and senior author of the study.

“Our study demonstrates a critical role for mitochondria-lysosome contact sites to maintain the health of peripheral nerves. Moreover, defects in this important pathway have been recently linked to the pathogenesis of multiple neurodegenerative diseases, including Parkinson’s disease and lysosomal storage disorders,” said Menichella, who is also an associate professor of Pharmacology.
According to the authors, next steps include understanding how defects in mitochondria-lysosome contact site dynamics and function contribute to the degeneration of peripheral neurons, uncovering new roles for mitochondria-lysosome contact sites, and identifying new pathways for how mitochondria and lysosomes interact with other organelles to maintain neuronal health and contribute to additional neurological disorders.
“Together, this work provides important insights into mitochondria-lysosome contact site regulation in peripheral neuropathy and has important consequences for advancing the fields of organelle contact site biology and neurodegeneration,” Menichella said.
Nirupa Doris Jayaraj, senior research associate in the Meninchella laboratory, was co-lead author of the study. Co-authors include Dongjun Ren, a postdoctoral fellow in the Department of Pharmacology; Tayler Belton, a research technologist in the Wong laboratory; George Shum, a research technologist in the Wong lab; Hannah Ball, a student in the Driskill Graduate Program in Life Sciences (DGP) program and a member of the Wong lab; and Dimitri Krainc, MD, chair and the Aaron Montgomery Ward Professor of Neurology and director of the Simpson Querrey Center for Neurogenetics.
This work was supported by National Institutes of Health grants NINDS R00 NS109252, NINDS Diversity Research Supplement 3R00NS109252-04S2, NINDS R24 NS098523 and R37 NS054154, R01 NS104295-0, and NIH HEAL initiative supplement R01 NS104295-01 and R01 AR077691-01.