

A novel cellular pathway regulates DNA damage and structural changes in cardiomyocytes, which contributes to the development of cardiac hypertrophy, according to a Northwestern Medicine study published in Circulation.
The findings also suggest inhibiting DNA damage could be an effective therapeutic strategy for treating cardiac hypertrophy, according to Hossein Ardehali, MD, PhD, the Thomas D. Spies Professor of Cardiac Metabolism and senior author of the study.
Proper nuclear organization, or the even distribution of DNA within the nucleus, helps cardiomyocytes regulate the contraction of the heart muscle and, in turn, reduce the risk of cardiovascular disease.
Previous work suggests that DNA damage in cardiomyocytes plays a significant role in cardiac hypertrophy — when the walls of the heart’s left ventricle thicken, making it difficult for the heart to pump blood. The mechanisms driving this process, which could serve as a therapeutic target, however, have not been well defined.
It has also been established that the AMPK (AMP-activated protein kinase) protein family regulates cell metabolism and response to DNA damage, as seen in cardiac hypertrophy.

In the current study, Ardehali’s team examined the role of a member of the AMPK protein family, the Snf-1 related kinase or SNRK, in DNA damage. Ardehali’s lab had previously discovered that SNRK improves mitochondrial efficiency, or energy production, in cardiomyocytes.
In SNRK-knockout mouse models, the investigators observed an exaggerated increase in heart size, which is frequently seen in patients with high blood pressure and diabetes, as well as DNA double-strand breaks in response to an increase in blood pressure, according to Ardehali.
“We wanted to see if the increase in DNA damage is the mechanism for the cardiac hypertrophy in these mice and understand the mechanism that is responsible for this phenotype,” said Ardehali, who is also a professor of Pharmacology and director of the Center for Molecular Cardiology.
To confirm their findings, the investigators used a process called phosphoproteomics to identify which proteins are altered by SNRK and discovered that the Destrin (DSTN) protein binds to SNRK, which reverses excess DNA damage, changes in nuclear organization and cellular hypertrophy, or the increase of cell size.
DSTN is a member of F-actin depolymerizing factor proteins that binds to and depolymerize F-actin. Ultimately, removing DSTN in SNRK-knockout mice increased this actin depolymerization in the cardiomyocytes, according to Ardehali.
“Nuclear morphology and the DNA damage go hand in hand, and actin is needed for that process,” Ardehali said.
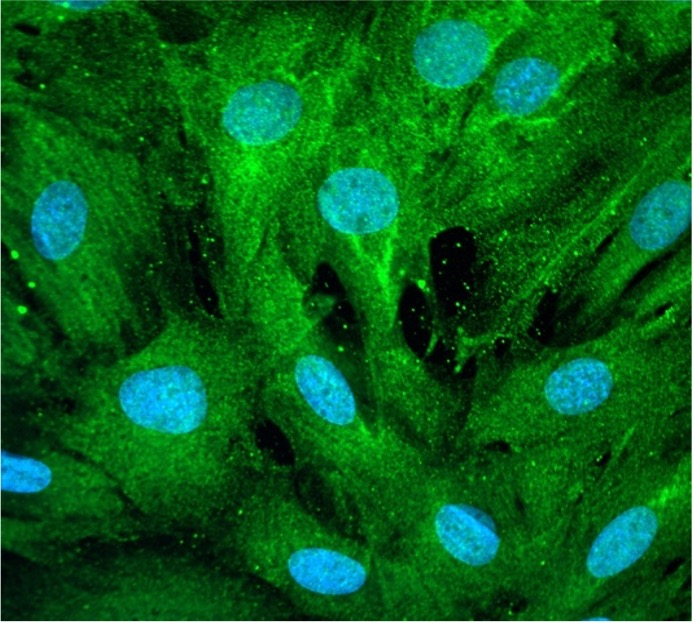
Lastly, the investigators administered a pharmacological stabilizer of F-actin to the SNRK-knockout mice, which led to an inhibition of DNA damage and reduced nuclear morphology in cardiomyocytes.
“We are hoping that in the future, we can use this drug to prevent cardiac hypertrophy. This is a novel approach to cardiac hypertrophy,” Ardehali said. “People have looked at so many different transcription factors, so many processes, including processes that are involving calcium handling for cardiac hypertrophy, but this is a novel approach, and we want to hopefully see whether or not we can reverse this process in the future in large animal models and in humans.”
Co-first authors of the study include Yuki Tatekoshi, MD, PhD, a postdoctoral fellow in the Ardehali laboratory, Paulina Stanczyk, PhD, a former postdoctoral fellow in the Ardehali laboratory, and Jason Shapiro, a student in the Medical Scientist Training Program (MSTP).
Co-authors include Matthew Schipma, PhD, research assistant professor of Biochemistry and Molecular Genetics, and Adam DeJesus, a student in the Medical Scientist Training Program (MSTP).
Ardehali is a member of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University.
This work was supported by National Heart, Lung, and Blood Institute grants HL127646, HL140973, HL138982 and HL140927, and the Leducq Network.





