

Northwestern Medicine investigators have discovered that mitochondria regulate essential cellular signaling for the development of epithelial cells in the lungs, cells which are crucial for the exchange of oxygen and carbon dioxide to avoid respiratory failure, according to findings published in Nature.
“Over the past twenty years, we have argued that mitochondria do more than just produce ATP; they control cell state and function by generating signals, including metabolites, to control physiology. If these signals are disturbed, disease can result. This study is a compelling demonstration of that principle and highlights the role of mitochondrial signaling in determining cell fate,” said Navdeep Chandel, PhD, the David W. Cugell, MD, Professor of Medicine in the Division of Pulmonary and Critical Care and of Biochemistry and Molecular Genetics and senior author of the study.
SeungHye Han, MD, MPH, assistant professor of Medicine in the Division of Pulmonary and Critical Care, was lead author of the study.
Mitochondria are widely known as cellular ‘powerhouses’ for their role in producing ATP, the source of energy for cells, and it had been previously thought that mitochondrial malfunction causes disease solely due to decreased energy production for cells.

Chandel and Han’s most recent findings, however, illustrate that mitochondria have functions beyond ATP production, including controlling the cellular fate of alveolar epithelial type 2 cells, which serve partially as stem cells and also regulate gas exchange in the lungs.
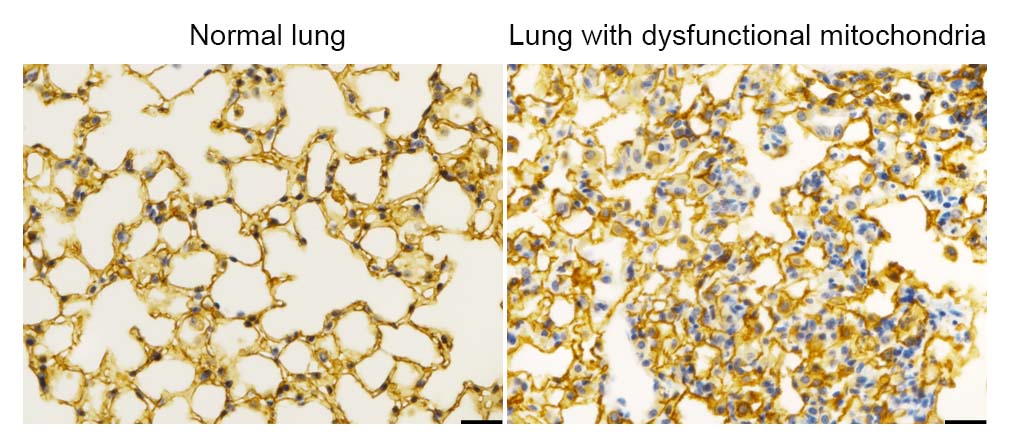
In the current study, the investigators used genetic knockout techniques to delete a mitochondrial electron transport chain complex I subunit, called Ndufs2, in lung epithelial cells in gestational mice. Next, using single-cell RNA sequencing, the team observed that mitochondria involve an integrated stress response (ISR), a cellular function that’s activated to cope with metabolic stress or proteotoxicity, or the accumulation of misfolded proteins.
When mitochondria malfunctioned, the ISR increased activation and inhibited the differentiation of alveolar epithelial cells in post-natal mice, ultimately leading to respiratory failure in the mice. Interestingly, the team also noted that the cells without functional mitochondria did not die, but instead remained in a state of stalled cell differentiation, which is commonly observed in a variety of lung diseases.
“We’re now examining the possibility that mitochondrial ISR signaling could be disrupted during lung repair after injury resulting in lung diseases like pulmonary fibrosis or prolonged viral pneumonia. If this is true, it could open new avenues for treatments that target mitochondria-dependent ISR signaling in diseases involving lung damage and repair,” Han said.
Co-authors of the study include Ram Chakrabarty, a student in the Driskill Graduate Program in Life Sciences (DGP); Laura Dada, PhD, research associate professor of Medicine in the Division of Pulmonary and Critical Care; Colleen Reczek, PhD, research assistant professor of Medicine in the Division of Pulmonary and Critical Care; Cara Gottardi, PhD, associate professor of Medicine in the Division of Pulmonary and Critical Care ; Mariana Herreria, PhD, a postdoctoral fellow in Division of Pulmonary and Critical Care; and Scott Budinger, MD, chief of Pulmonary and Critical Care in the Department of Medicine and the Ernest S. Bazley Professor of Airway Diseases.
Chandel, Gottardi and Budinger are members of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University.
This work was supported by the National Institutes for Health grants R35CA197532, P01HL071643, P01AG049665, K08HL143138, R01HL134800 and T32HL076139; American Heart Association Career Development Award (19CDA34630070), the Parker B. Francis Fellowship, the Doris Duke Charitable Foundation/Walder Foundation and Feinberg School of Medicine COVID-19 Fund to Retain Clinician Scientists; the Veterans Administration grant CX001777; and the Northwestern University Pulmonary and Critical Care Cugell fellowship.





