Northwestern Medicine investigators have discovered a novel mechanism linking an intercellular adhesion molecule with genetic inflammatory skin diseases, according to findings published in the Journal of Clinical Investigation.
The study, led by Kathleen Green, PhD, the Joseph L. Mayberry, Sr., Professor of Pathology and Toxicology and a professor of Dermatology, suggests that Desmoglein-1 (Dsg1) deficiency results in an inflammatory gene signature that is seen in patients diagnosed with skin diseases such as psoriasis and may be a promising therapeutic target.
Dsg1 is a cadherin, a cell-to-cell adhesion molecule and key component of the desmosome, a complex that helps tissues withstand mechanical and environmental stressors. Dsg1 is expressed exclusively within the upper layers of the skin’s most outer layer, the epidermis.
Previous work suggests that mutations in Dsg1 and ultimately its loss of function are associated with chronic inflammation and the manifestation of skin lesions and allergies. However, the precise intracellular mechanisms that cause this deficiency remained unknown.

To uncover these mechanisms, the investigators used gene editing methods to delete Dsg1 from mice genomes. The gene expression profiles of the mice were then compared to the profiles of patients with severe dermatitis, multiple allergies and metabolic wasting (SAM) syndrome, a rare, genetic skin disease caused by Dsg1 deficiency that results in a severe thickening of the skin and increased inflammation.
The team found similarities between the genetic profiles of the mice and patients, specifically a shared IL-17/IL-23 inflammatory signature and impaired intercellular adhesion caused by the loss of Dsg1 at the surface of cells in the epidermis. IL-17 is secreted by T-cells involved in the body’s inflammatory response. This inflammatory signature was also similar to that seen in patients with psoriasis, the authors found.
Further demonstrating the relevance of their findings in vivo, two Dsg1-deficient patients with SAM syndrome were treated with ustekinumab, an immunosuppressive drug commonly prescribed to treat psoriasis. The drug inhibits IL-12/IL-23 cytokine activity, which is involved in the body’s inflammatory response. After 10 months of treatment, both patients experienced significant improvement of pre-existing skin lesions.
“We’ve only begun to scratch the surface and now we’re interested in how much Dsg1 might be playing a role in other skin diseases,” said Lisa Godsel, ’97 PhD, research assistant professor of Pathology, of Dermatology and co-lead author of the study.
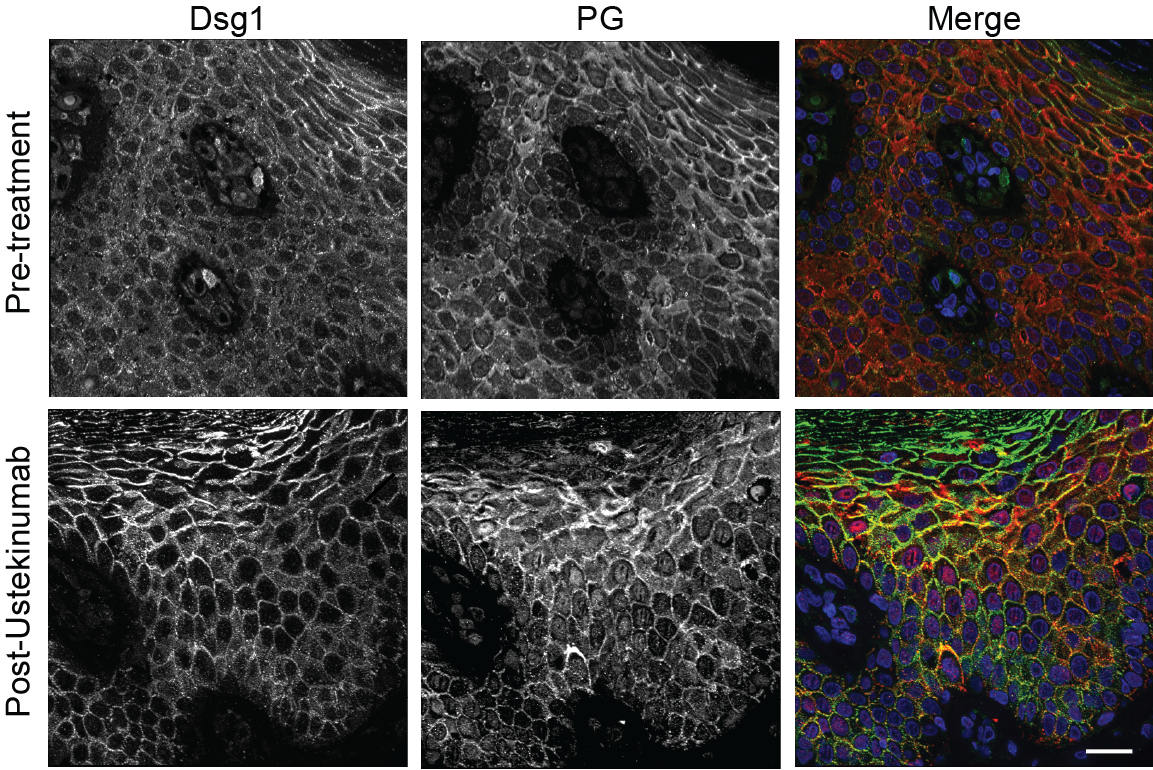
At the very least, Dsg1 expression could serve as a biomarker that can be used to improve treatment of genetic skin diseases, Godsel added. Furthermore, a topical treatment that can help increase the expression of Dsg1 in the skin would be a game changer in treating these diseases.
“If we can actually improve Dsg1 expression, we might be able to target all these diseases that have a Dsg1 decrease,” Green said.
Quinn Roth-Carter, PhD, a postdoctoral fellow in the Green laboratory, was co-lead author of the study. Co-authors include Joshua Broussard, PhD, research assistant professor of Pathology and of Dermatology; Marihan Hegazy, a student in the Driskill Graduate Program in Life Sciences (DGP); Jodi Lynn Johnson, PhD, research assistant professor of Pathology and of Dermatology; Lynn Doglio, PhD, research associate professor of Pharmacology; Rajeshwar Awatramani, PhD, professor in the Ken and Ruth Davee Department of Neurology Division of Movement Disorders; and Xiaomin Bao, PhD, assistant professor of Dermatology.
Green, Broussard, Johnson, Awatramani and Bao are members of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University, and Green is the associate director for Basic Sciences Research at the Lurie Cancer Center.
This work was supported by the National Institutes of Health (NIH) National Institute of Arthritis and Musculoskeletal and Skin Diseases grants R01 AR041836 and R01 AR075015, NIH grant R37 AR43380, National Cancer Institute grant R01 CA228196, a Leo Foundation Grant, NIH T32 Training grants CA070085 and CA009560, a JL Mayberry endowment, and NIH grant K01 AR075087.