An inflammatory pathway previously believed to protect the heart after myocardial infarction actually causes damage, according to a Northwestern Medicine study published in the Journal of Clinical of Investigation.
Inhibiting this inflammatory pathway reduced heart attack-induced damage in experimental models, a proof of concept that suggests this could be an effective therapy for patients after a heart attack, according to Edward Thorp, PhD, associate professor of Pathology in the Division of Experimental Pathology and senior author of the study.
“If patients could take this drug after acute myocardial infarction, we could potentially improve heart repair and therefore reduce their progression to heart failure,” said Thorp, who is also a professor of Pediatrics.
Cardiovascular disease, heart attacks and heart failure are leading causes of morbidity and mortality in the United States. One significant contributor to heart failure is permanent heart damage after heart attack: Cardiomyocytes, the muscle cells that make up most of the heart, do not regenerate in adulthood and often die in heart attacks, leaving large swaths of scar tissue across the heart.
One contributor to this damage is excessive inflammation caused by reperfusion therapy often given to heart attack patients when they reach the hospital. The sudden influx of oxygen and blood that results from opening blocked arteries and restoring blood flow can shock the system, according to Thorp.
Scientists have worked to understand the inflammatory mechanisms after heart attacks, and reduce this self-inflicted damage. One receptor on macrophages, AXL, an all-purpose immune cell, was previously implicated in suppressing inflammation.
Thorp and his collaborators initially believed that enhancing the function of AXL could help the heart heal better by suppressing inflammation, but surprisingly, they found the opposite. Inhibiting this molecule in mouse models of heart attack actually reduced damage to the heart.
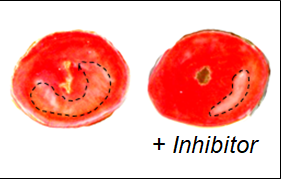
“This was a surprise, but it fueled excitement and additional experiments for the observation that challenged not only our own assumptions but also the prevailing role for this receptor in the published literature,” said Matthew DeBerge, PhD, research assistant professor of Pathology and lead author of the study.
The scientists traced phenomenon, and found that the receptor increases the metabolism of macrophages, in turn producing a pro-inflammatory cytokine called IL1-beta. This two-step process was previously unknown, and helps explain why the AXL receptor can cause damaging inflammation, according to Thorp.
“This requires a shift in conceptual thinking,” Thorp said. “This receptor, which for all intents and purposes was predicted to be cardio-protective, is actually maladaptive and causes cardiac injury.”
There is evidence that the AXL receptor is upregulated during heart attacks and heart failures, and Thorp said he believes it could be used as a biomarker, highlighting patients who would benefit from an AXL inhibitor immediately after heart attack.
“These drugs are tricky, because they suppress the immune system, which when used in chronic conditions can open people up to opportunistic infections,” Thorp said. “But this inflammation is in such a tight window, I think we could administer the therapy just to target this post-myocardial infarction inflammation and then stop the therapy.”
In the future, the authors said they are eager to explore how the AXL receptor regulates macrophage metabolism and are actively exploring opportunities for human clinical trials testing the effectiveness of AXL inhibitors in reducing post-heart attack damage.
“Our study targeting AXL after heart attack demonstrates that we can inhibit inflammatory macrophage function, while sparing macrophage function that is beneficial for tissue repair to improve healing and limit the progression to heart failure,” DeBerge said.
Lisa Wilsbacher, ’03 MD, PhD, assistant professor of Medicine in the Division of Cardiology and of Pharmacology, was a co-author of the study.
This work was supported by American Heart Association grant CDA24110032 and National Institutes of Health grants F32HL127958, R01HL122309, R01HL139812, R35HL145228 and R01HL127464.