By studying a genetic mutation in patients with a rare Parkinson’s-like disorder, Northwestern Medicine scientists are working to to understand the mechanisms behind the more common form of the disease.
In a recent study, Dimitri Krainc, MD, PhD, Aaron Montgomery Ward Professor and chair of the Ken and Ruth Davee Department of Neurology, and Taiji Tsunemi, MD, PhD, research assistant professor in Neurology, studied a mutation in the PARK9 gene, which is found in patients with Kufor-Rakeb syndrome. The disorder is characterized by juvenile-onset symptoms similar to Parkinson’s disease, including movement abnormalities and dementia.
Drs. Krainc and Tsunemi explored how the PARK9 mutation contributes to abnormal accumulation of α-synuclein, a protein that facilitates communication between neurons.
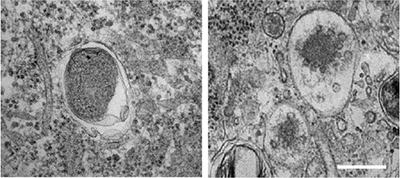
The answer involved cellular recycling factories in the brain called lysosomes, which are organelles responsible for removing cell debris. When they malfunction, α-synuclein builds up in cells instead of being properly disposed, contributing to neurodegeneration and symptoms of parkinsonism.

“We found that lysosomes do not work properly in patients with PARK9 mutation, suggesting that PARK9 plays a role on normal function of lysosomes,” Dr. Krainc said.
The scientists also found that the PARK9 mutation also impairs exosomes, cellular vesicles that release α-synuclein from one cell to those cells neighboring it.
Patients with Kufor-Rakeb syndrome respond well to the same symptomatic drug treatment that patients with Parkinson’s disease take. This all implies that the underlying mechanisms of both conditions are similar.
“We study this rare disease because we know the specific gene that is abnormal,” said Dr. Krainc, who is also director of the Center for Rare Neurological Diseases. “Clear disease mechanisms and targets for the rare genetic disorders provide a window that can help us establish a platform to develop new drugs for both the rare and common disorders.”
The findings of this study, published in the Journal of Neuroscience, could apply to more frequent forms of Parkinson’s disease that don’t have a clear genetic root.
The scientists are currently investigating whether lysosomal function is also abnormal in patients with Parkinson’s disease, to see if their findings about the rare disease correlate to more common forms.
“If that’s the case, we can try to upregulate the activity of this gene and the protein, to improve the recycling capacity of neurons so they can digest accumulated debris in much more efficient way,” Dr. Krainc said.
This work was supported by National Institutes of Health (NIH) grant R01NS076054.