Mitochondria work like a power plant inside cells to regulate energy supply. When mitochondrial health is targeted and destroyed a lot can go wrong, especially when the attacker is associated with fatal diseases such as amyotrophic lateral sclerosis (ALS).
In a new study, Northwestern Medicine scientists showed direct mitochondrial destruction by a protein from the FUS gene, one of the genes associated with ALS as well as frontotemporal lobar degeneration (FTLD) and other neurodegenerative diseases.
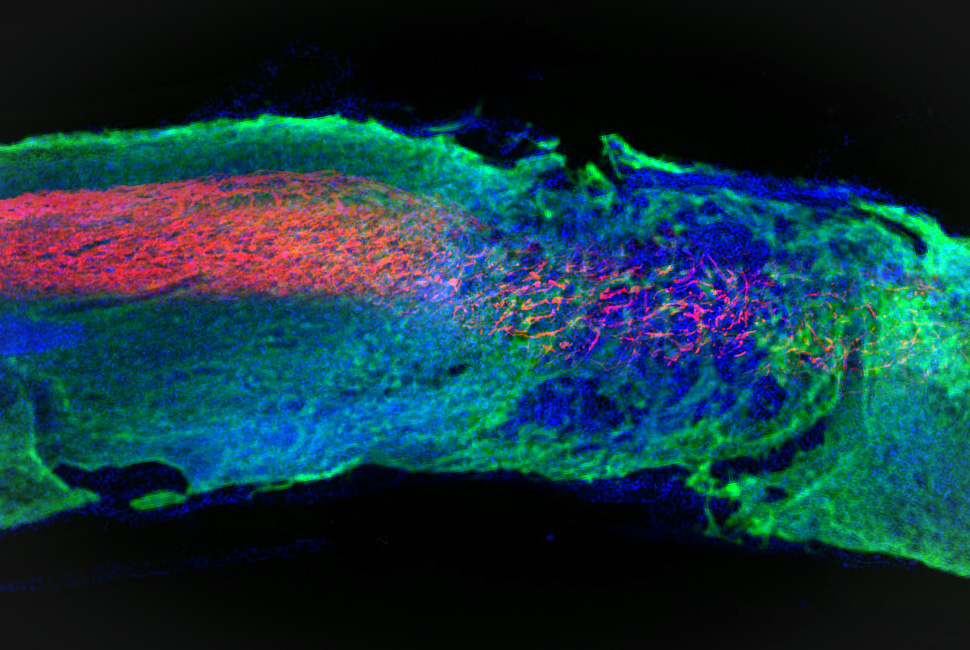
Mitochondrial damage done by the nuclear FUS protein was found in a fruit fly model and in tissue of patients with neurodegenerative diseases. This damage seems to occur early on in disease progression, making mitochondrial health an emerging area of interest for scientists developing early diagnostic tools and therapies for neurodegenerative diseases.
This previously unknown and unexpected discovery about the destructive aspect of this protein in mitochondria was published September 3 in PLOS Genetics.
“FUS mitochondrial localization is toxic to neurons, contributing to neurodegeneration,” said Jane Wu, MD, PhD, the Dr. Charles L. Mix Research Professor of Neurology and Psychiatry, who led the study. “We are actively working on developing new diagnostic tools based on mitochondrial damage for neurological diseases. We hope that earlier diagnosis before the disease progression to an irreversible stage is critical for successful treatment.”
Dr. Wu, a professor of Neurology, and her team have spent the past two decades studying critical players in dementia and motor neuron diseases. Wu hopes the development of early diagnostic tools will help diagnose these diseases before they become irreversible. Early diagnosis will allow scientists to develop therapeutic approaches that enhance patients’ quality of life and possibly reduce fatalities.
This work is the result of multi-group interdisciplinary collaboration among scientists from Feinberg and the Chinese Academy of Sciences and Nanjing University.
The Chinese team was funded by Ministry of Science and Technology- China 973 Projects (2010CB529603 & 2013CB917803, 2010CB529603) and NSFC grant (91132710). Dr. Wu and her team were supported by the ALS Therapy Alliance and National Institutes of Health grants NIA P30 AG13854, R56NS074763, U54 CA151880 and RO1AG033004.
Read more about this study in a Q&A with Dr. Wu:
Q: What led your team to study FUS and mitochondrial damage?
A: We have been interested in understanding molecular function of RNA binding proteins in the nervous system. We had been pursuing another project using brute force biochemical purification and mass-spectrometry analyses to identify new mitochondrion-associated proteins when the study of FUS mutations in ALS patients was published.
Because we identified FUS as a candidate mitochondrion-associated protein, we began to investigate the role of FUS in affecting mitochondrial health. The observation that FUS-positive pathological lesions were found in patients with and without FUS gene mutations led us to examine the effects of expression of either wild-type or ALS-mutant FUS in our cellular models and animal models of FUS proteinopathies.
Q: What are the most important findings in this study?
A: Our data provide the first evidence of direct mitochondrial targeting by a nuclear protein associated with neurodegeneration. Our study suggests that mitochondrial impairment may represent a critical event in different forms of FUS-proteinopathies and a common pathological feature for both ALS-FUS and FTLD-FUS. Our work offers a potential explanation for the highly heterogeneous nature and complex genetic presentation of different forms of FUS-proteinopathies. Our study also suggests that mitochondrial damage may be a target in future development of diagnostic and therapeutic tools for FUS-proteinopathies, a group of devastating neurodegenerative diseases.
Q: Why do you use the fruit fly model?
A: Fruit flies (drosophila melanogaster) have been studied for decades as a great tool for genetic research. The vast majority of the drosophila genome has been sequenced and systematically analyzed. The majority of drosophila protein-coding genes are conserved in the human genome. With a much simpler nervous system yet powerful genetics and a diverse range of functional assays established in drosophila, it has become a great animal model system for studying a diverse range of human diseases from neurodegenerative disorders to cancer.
Our team is one of the first to establish a drosophila model for studying FTLD and ALS associated with RNA binding proteins including both FUS and TDP-43 (Li et al, 2010: Chen et al, 2011). Using these Drosophila models for FTLD and ALS, we have made significant contributions to understanding their pathogenetic mechanisms.
Q: How close are you and other scientists to developing better diagnostic and therapeutic options for people with diseases such as ALS?
A: Our study sheds new light on the pathogenic mechanism of FTLD and ALS associated with FUS pathology. We are actively working on developing new diagnostic tools based on mitochondrial damage for neurological diseases. We hope that earlier diagnosis before the disease progression to an irreversible stage is critical for successful treatment.
Q: Why do you study ALS and FTLD?
A: FTLD is a common neurologic disease, overlapping clinically and pathologically with other common forms of dementia such as Alzheimer’s disease. ALS represents a spectrum of adult-onset neurodegenerative diseases. Although rare, patients affected by ALS often become symptomatic at 40 to 60 years of age, with disease progressing quickly with a high fatality rate.
Genetic studies in the past two decades have begun to uncover the underlying genetic defects among these patients. I believe that vigorous studies using interdisciplinary approaches will lead to not only better understanding of the underlying pathogenic mechanisms, but also identification of molecular targets and biomarkers for early diagnosis of these patients. Development of early diagnostic tools that enable us to diagnose these diseases before they become irreversible will allow us to develop therapeutic approaches that will enhance a patient’s quality of life and possibly reduce the fatality of these debilitating neurodegenerative diseases.