Northwestern Medicine scientists have identified the consequences of contact between the mitochondria and the lysosome in the axons and synapses of human neurons, according to a study published in Nature Communications.
Prolonged contact, observed in neurons modeling Parkinson’s disease, caused aberrant distribution of mitochondria, contributing to the disease’s neuronal dysfunction. Investigators traced this phenomenon to mutations in the lysosomal enzyme β-glucocerebrosidase (GBA), according to Dimitri Krainc, MD, PhD, chair and the Aaron Montgomery Ward Professor of Neurology and senior author of the study.
“Our study is the first to demonstrate that mitochondria-lysosome contact sites dynamically form in mammalian neurons and are misregulated in Parkinson’s disease, leading to downstream pathogenic events,” said Krainc, who is also director of the Simpson Querrey Center for Neurogenetics.
Parkinson’s disease is among the most common neurodegenerative disorders, in which dopaminergic neurons in the substantia nigra brain region begin to die, causing loss of motor control. There are currently no disease-modifying therapies for Parkinson’s disease, so studying key pathways involved in pathogenesis is vital to identify therapeutic targets, Krainc said.
Recent studies from the Krainc laboratory — published in Nature and Science — have highlighted the role of mitochondrial and lysosomal dysfunction in Parkinson’s disease, suggesting that direct signaling between these two organelles may represent an important component of the disease.
In the current study, Soojin Kim, a fifth-year student in the Northwestern University Interdepartmental Neuroscience (NUIN) Program and lead author of the study, used super-resolution time-lapse microscopy to observe mitochondria and lysosomes in contact with each other throughout the dendrites and axons of nerve cells from patients with Parkinson’s disease. Importantly, she also discovered that neurons harboring loss-of-function mutations in GBA — considered one of the most serious risk factors for Parkinson’s disease — also significantly prolonged mitochondria-lysosome contact compared to normal neurons.
“This suggests that misregulation of mitochondria-lysosome contacts is an important step of disease pathogenesis,” Krainc said.
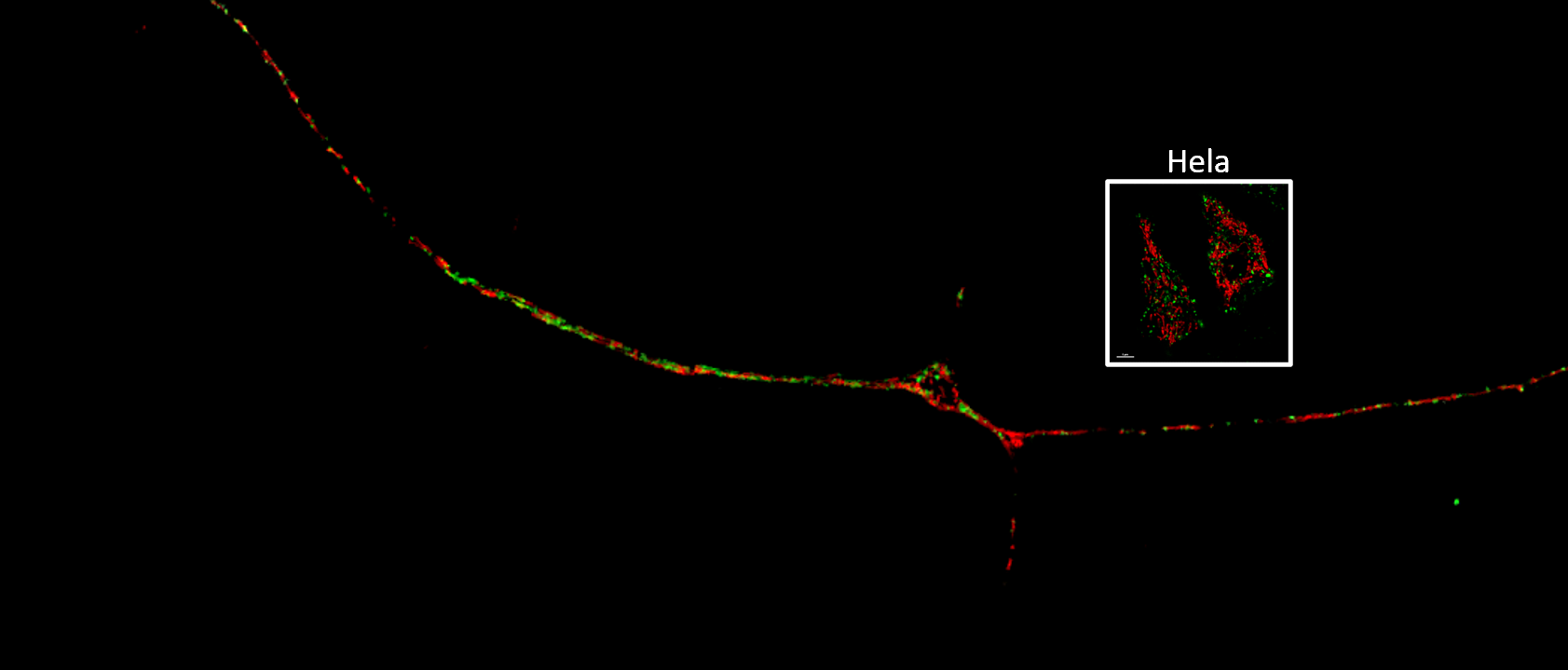
Digging deeper, the investigators found that mutant GBA in patient neurons alters important mediators of mitochondrial-lysosome contact, resulting in fewer mitochondria in the axons of neurons and lowered levels of ATP, the source of energy for cells.
Surprisingly, these downstream effects were rescued by upregulating the TBC1D15 protein, which is involved in mitochondria-lysosome tethering.
“When we knockdown TBC1D15 in healthy neurons, we see the phenotypes we’ve observed in diseased neurons,” Kim said. “When we overexpress TBC1D15 in diseased neurons, we could partially rescue downstream pathological phenotypes.”
These findings highlight mitochondrial-lysosomal contacts as an upstream regulator of mitochondrial function and the complex dynamics driving neurodegeneration in Parkinson’s disease, according to Krainc.
“Understanding the role and regulation of inter-organelle contacts has critical implications for multiple neurodegenerative diseases, and offers an important previously unstudied angle for understanding the convergence of mitochondrial and lysosomal dysfunction in Parkinson’s disease etiology,” Krainc said.
This study was supported by National Institutes of Health National Institute of Neurological Disorders and Stroke grants F32 NS101778, K99 NS109252, R01 NS076054 and R37NS096241, and a Warren Alpert Distinguished Scholars Fellowship Award.