The strength of neuron-to-neuron connections does not govern the spread of Parkinson’s disease in the brain, according to a Northwestern Medicine study published in Science Advances.

This upends an emerging theory about the underlying mechanisms of Parkinson’s disease. Learning more about the pattern of spread could improve future therapies for the degenerative nervous system disorder, according to D. James Surmeier, PhD, chair and Nathan Smith Davis Professor of Physiology and senior author of the study.
“There is something besides just the number of synaptic connections that is dictating spread and severity of Parkinson’s disease,” Surmeier said. “Exactly what, we can’t yet say. But, figuring out what make neurons more or less vulnerable will give us important clues about how to stop the spread.”
Intracellular clumps of debris — misfolded proteins, damaged lipids and pieces of partly digested organelles — are one of the hallmarks of Parkinson’s disease. One major component of these clumps, called Lewy bodies, is a misfolded protein called alpha-synuclein.
One hypothesis is that misfolded alpha-synuclein spreads from neuron to neuron through synaptic connections, causing neurons to stop working properly and eventually die. Recent studies have raised the possibility that the main determinant of spread is the number of synaptic connections between neurons — but Surmeier and his colleagues were skeptical, he said.
“If you look at the distribution of pathology in the Parkinson’s brain, it doesn’t conform to any strict synaptic connectome,” Surmeier said.
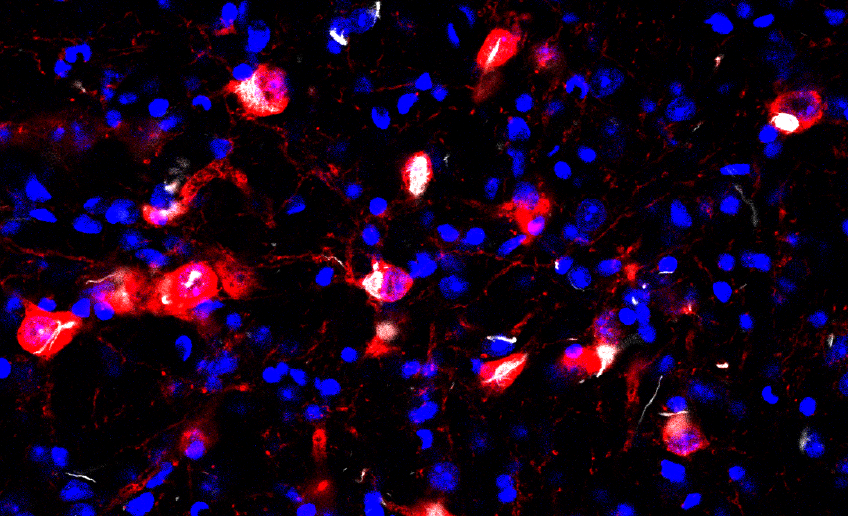
Testing this theory in mouse models of Parkinson’s disease, the investigators injected a misfolded, pathological form of alpha-synuclein protein into a specific region of the brainstem. This region, an area called the pedunculopontine nucleus, often manifests Lewy pathology in humans.
Then, they traced where the misfolded alpha-synuclein traveled throughout the brainstem. The first surprising observation was that the misfolded alpha-synuclein was only taken up by one of three types of neuron: neurons that use acetylcholine as a transmitter.
“This was encouraging because it is precisely these cholinergic neurons that have Lewy pathology in Parkinson’s disease,” Surmeier said.
Next, they compared the inputs and outputs of these cholinergic neurons with the spread of the misfolded alpha-synuclein. Importantly, the spread of alpha-synuclein pathology was almost exclusive to neurons making synaptic connections onto the cholinergic neurons.
“The spread of alpha-synuclein pathology was retrograde — away from an in ‘infected’ neuron,” Surmeier said.
The investigators found that nearly all of the neurons that made synaptic connections with the ‘infected’ cholinergic neurons got ‘infected’ themselves. However, contrary to current thinking, the severity of the pathology was not related to the strength or number of synaptic contacts. Moreover, in many regions of the brain, the pathology went away with time, suggesting that no permanent damage had been done.
“It didn’t matter how many connections there were between two regions,” Surmeier said. “There is something else that determines the chances of spreading.”
One possibility is that activity at synapses controls the spreading process. Another possibility, which is preferred by Surmeier, is that some cells express surface proteins that act as “Trojan horses” to bring alpha-synuclein into the cell. If this were the case, those surface proteins could be targeted with drugs or antibodies to block transmission.
This kind of targeted strategy contrasts with current therapeutic efforts that attempt to use antibodies that bind to alpha-synuclein itself.
“This alpha-synuclein antibody strategy may be effective for slowing the spread of Lewy pathology, but it doesn’t take advantage of the insights obtained about the mechanisms of spread,” Surmeier said. “It’s an indiscriminate sledgehammer.”
Instead, identifying what exactly makes cells vulnerable to Lewy pathology could result in more targeted and effective treatments, according to Surmeier.
The study was the result of an international collaboration between the Surmeier laboratory, the Dawson laboratory at Johns Hopkins School of Medicine and the Oertel laboratory at Philipps University in Marburg, Germany. Martin Henrich, MD, Fanni Geibl, MD and Harini Lakshminarasimhan, PhD, members of the Surmeier laboratory, were lead author and a co-authors of the study, respectively.
This work was supported by the JPB Foundation, ParkinsonFonds Deutschland, the Charitable Hertie Foundation, the German Society for Parkinson and Movement Disorders, National Institutes of Health grants K01AG056841 and R01NS107318, American Parkinson’s Disease Association grant 90076052 and the Parkinson’s Foundation Stanley Fahn Junior Faculty Award PF-JFA-1933.