Northwestern Medicine scientists have discovered that a genetic mutation found in amyotrophic lateral sclerosis (ALS) patients disrupts the localization of proteins involved in RNA and protein metabolism and leads to the dysfunction and eventual degeneration of motor neurons, according to a study published in Neuron.
The findings, which shed light on the mechanisms and consequences of a defect in the gene, known as C9orf72, may aid the development of novel therapeutic interventions for patients with the neurodegenerative disease, which currently has no cure.

“Our study shows very interesting ways of how a neuron tries to defend itself from cellular stress inferred by the C9orf72 mutation. For instance, we found that motor neurons form nuclear invaginations that accumulate the necessary machinery required to adjust the level of protein and RNA metabolism,” said Juan Alberto Ortega, PhD, a postdoctoral fellow in the Kiskinis laboratory at the Les Turner ALS Center and co-first author of the study.
Previous work has found that this genetic mutation impairs the exchange of molecules between the nucleus and the cytosol, a process known as nucleocytoplasmic trafficking in which proteins and ribonucleic acid (RNA) move across the nuclear envelope of a cell.
“What we wanted to identify, however, was what are the proteins that are redistributed as a result of this trafficking defect,” said Evangelos Kiskinis, PhD, assistant professor in the Ken and Ruth Davee Department of Neurology and senior author of the study.
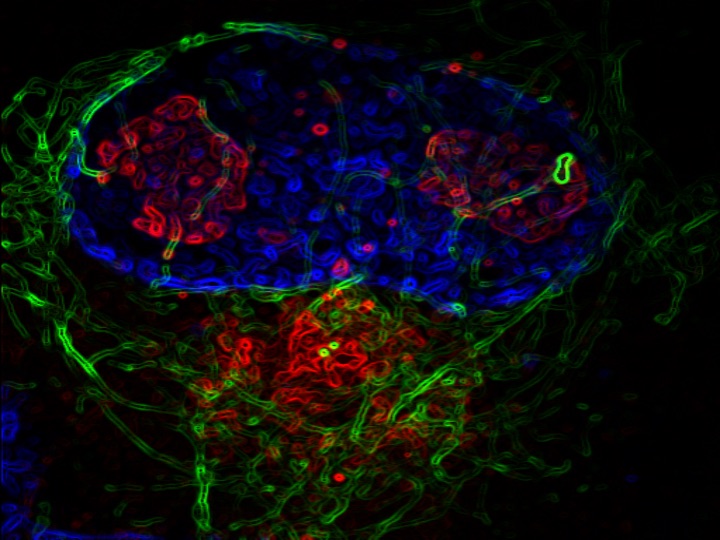
For the study, the scientists used subcellular fractionation — a method which involves the biochemical separation of the nucleus from the rest of the cell — and mass spectrometry to conduct subcellular proteomic screening in a cellular model of ALS.
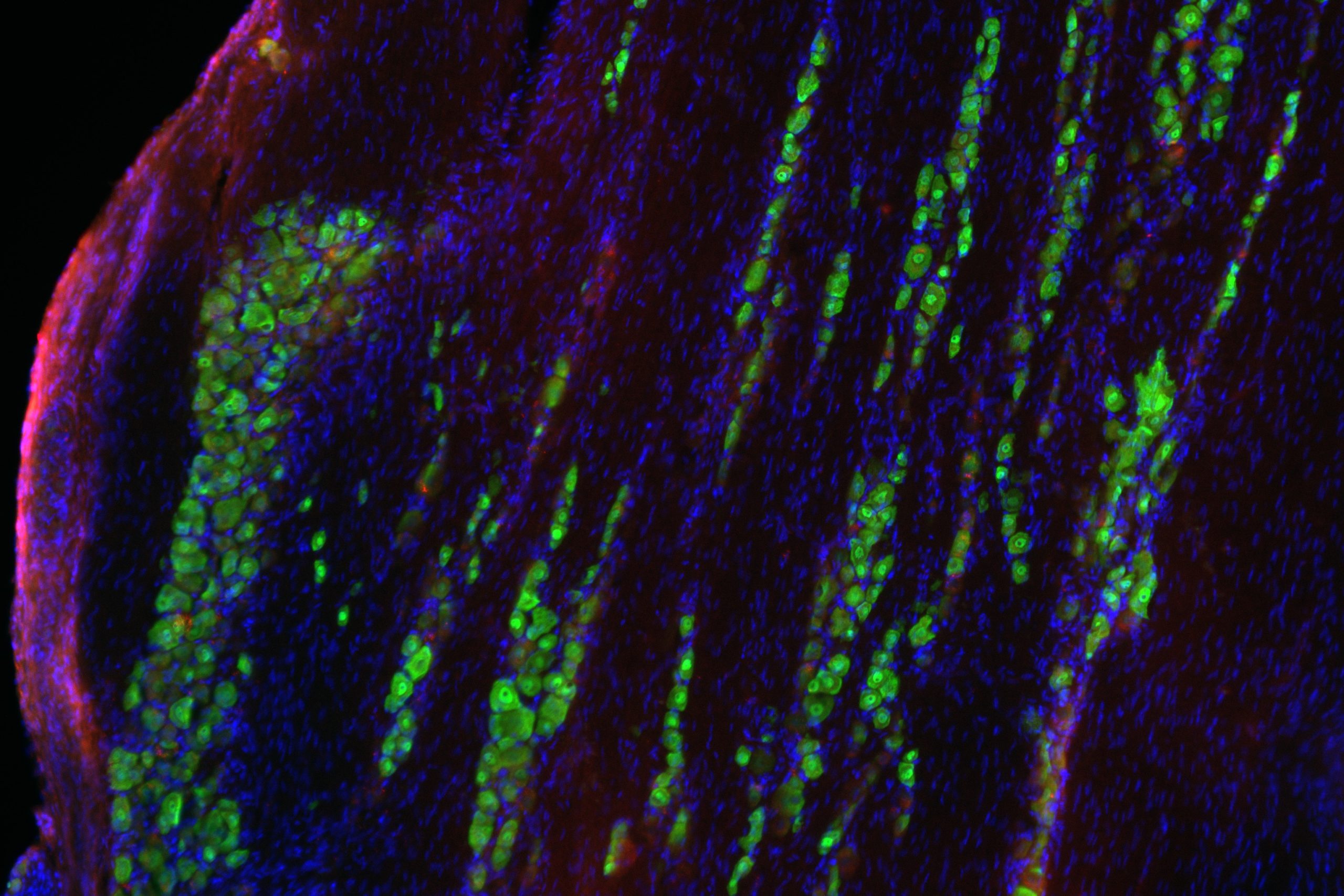
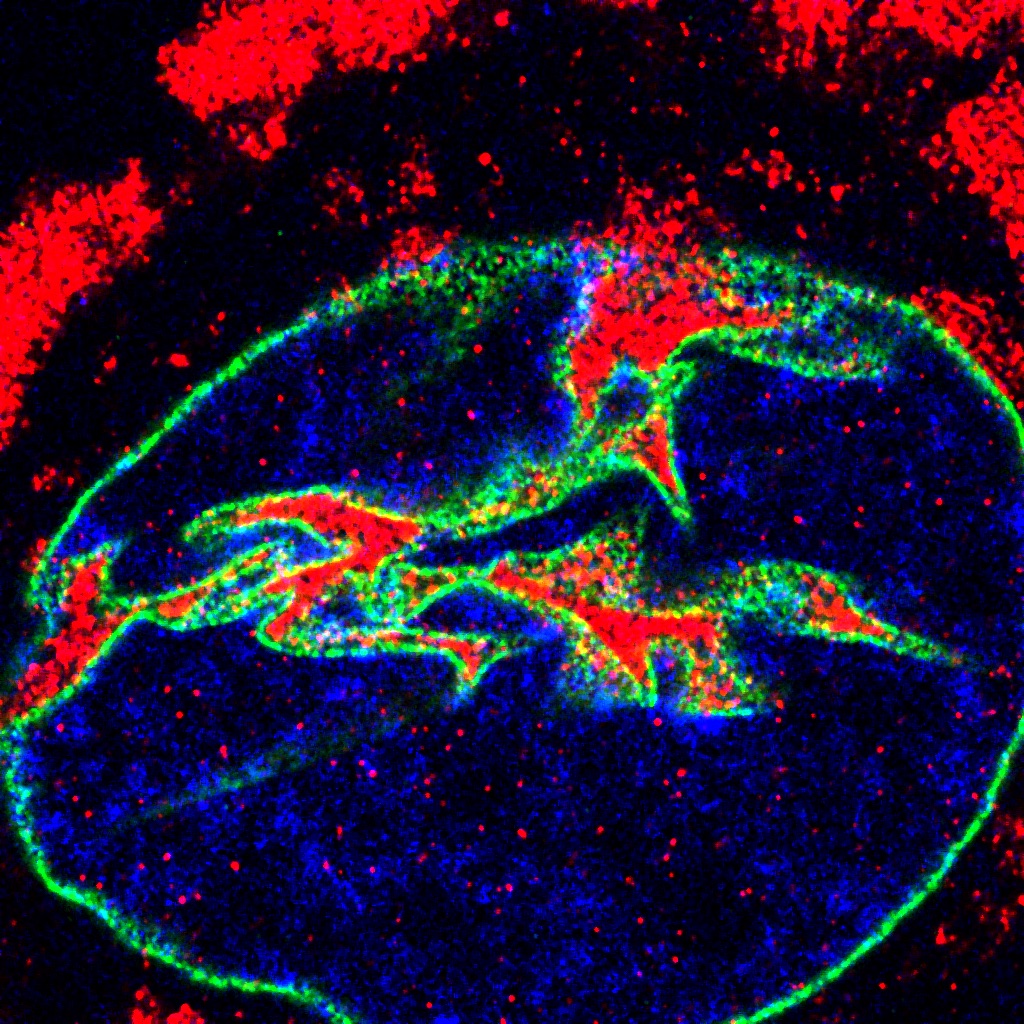
From this analysis, the scientists found that there is an imbalance of the nuclear and cytoplasmic proteome in the ALS cells, and this imbalance targets proteins involved in RNA and protein metabolism. Additionally — and unexpectedly — the team also found that the eRF1 protein, which is normally found in the cytosol of a cell, accumulates within nuclear invaginations in neurons and plays a major role in the pathophysiology of the disease.
Then, using in vitro and in vivo models of the protein, the scientists found that eRF1 shifts the balance between protein translation and RNA degradation through a quality control pathway called nonsense-mediated decay.
“Our specific protein of interest, eRF1, mediates the balance between translation and RNA degradation by nonsense mediated decay,” said Kiskinis, who is also a professor in the Division of Neuromuscular Disease and of Physiology. “What we found was that this protein accumulates within nuclear invaginations, and triggers a reduction in protein translation and an increase in the degradation of RNA molecules including the mutant C9orf72 transcript itself. To be able to visualize eRF1 within these invaginations we had to use expansion microscopy.”
According to Kiskinis, these findings shed light on a cell-intrinsic pathway that degrades the mutant RNA; finding a way to trigger that pathway could be therapeutically relevant.
“This study is exciting because, based on an unbiased approach, our findings pointed to a specific RNA degradation pathway, which may be a key underlying component of disease,” said Elizabeth Daley, a fifth year graduate student in the Northwestern University Interdepartmental Neuroscience Program (NUIN) and co-first author of the study.
Seed funding for the laboratory’s work on ALS was provided by the Les Turner ALS Foundation, while support by the New York Stem Cell Foundation, which recently awarded Kiskinis a New York Stem Cell Foundation – Robertson Investigator Award, will enable the continuous investigation of degenerative mechanisms in ALS.
This work was supported by the National Institutes of Health National Institute on Neurological Disorders (NINDS) and Stroke and National Institute on Aging grant R01NS104219, NIH and NINDS grants R21NS111248 and R21NS107761.