Northwestern Medicine scientists identified how genetic variants in the gene BIN1 can contribute to late-onset Alzheimer’s disease, according to a recent study published in Molecular Psychiatry.
A group of scientists led by Peter Penzes, PhD, the Ruth and Evelyn Dunbar Professor of Psychiatry and Behavioral Sciences, discovered that mutations in BIN1 can weaken synapses, the bridges between nerve cells. While dysfunctional synapses may not be enough to cause Alzheimer’s disease on their own, Penzes notes these findings point towards a multifaceted model of disease.
“If a patient has one mechanism of disease, like this gradual weakening of synapses, they might be more sensitive to other mechanisms, like amyloid protein buildup,” said Penzes, also director of the Center for Autism and Neurodevelopment and a professor of Physiology and of Psychiatry and Behavioral Sciences.
Little is known about the genetic causes of late-onset Alzheimer’s disease (LOAD), despite being the most common form of the disease. Until relatively recently, most research investigated the rarer but hereditary familial Alzheimer’s disease, Penzes explained, as it was easier to track and identify genes that were passed down through generations compared to the sporadic mutations found in late-onset disease.

This changed a decade ago, when a large genome-wide analysis identified 20 “risk” genes — including BIN1 — associated with LOAD. For the first time, scientists had a roadmap for further investigation, according to Penzes.
“These were new and obscure genes,” Penzes said. “How they contributed to disease, or affected neurons and the brain was not known.”
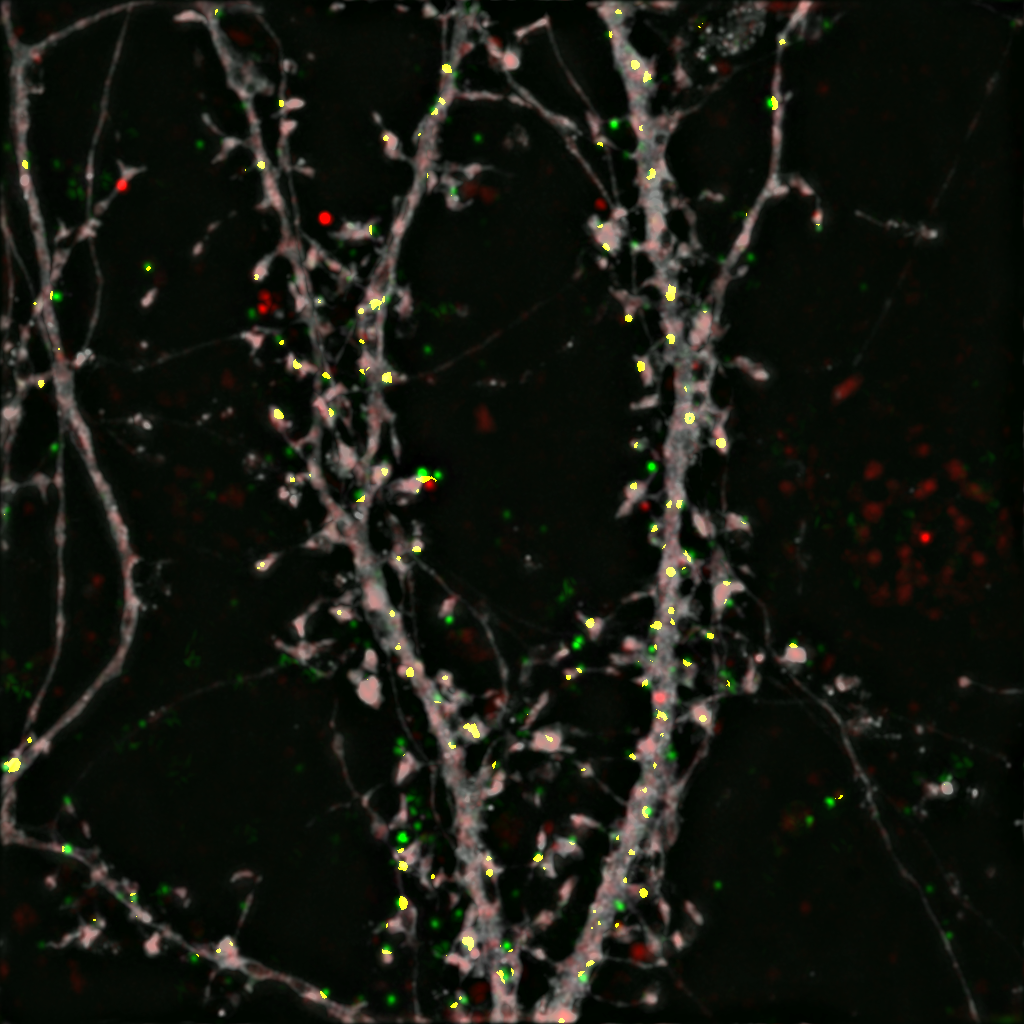
To find out, Penzes and his colleagues used structural illumination microscopy to peer inside the synapses of both healthy mouse neurons and those with BIN1 deleted, mimicking the mutation associated with LOAD.
With this new microscopy technology, the scientists could see where BIN1 proteins were active within the healthy cells, and found they were involved in transporting receptor proteins in and out of the synapse — a process called synaptic trafficking.
With BIN1 deleted, synaptic trafficking broke down, signaling that defective BIN1 could be the source of synaptic dysfunction in patients with LOAD, according to Penzes.
“Having less BIN1 over the long run could result in weaker neurotransmission,” Penzes said.
Penzes hopes these findings can help steer Alzheimer’s science away from the focus on familial Alzheimer’s and its primary mechanism of disease: buildup of harmful tau or amyloid proteins in the brain. The wealth of research into familial Alzheimer’s has resulted in an outsize focus on investigating and developing therapies for these symptoms, according to Penzes.
“The culture of the field was dominated by amyloid theory,” Penzes said.
However, the failure of amyloid-blocking drugs in clinical trials has caused the field to undergo self-reflection, Penzes said, and consider other mechanisms of disease. In fact, other LOAD-associated genes have similar gene expression patterns and functions as BIN1, raising the possibility that they too could be implicated in synaptic trafficking.
“It would be very interesting to see how these Alzheimer’s risk genes fit into synaptic regulation,” Penzes said. “We’re probably going to be at work for the next few decades, figuring out what all of those genes do and how they interact with each other.”
Britta Schurmann, PhD, and Daniel Bermingham, PhD, postdoctoral fellows, were the first authors of the study.
The study was supported by R01 grants MH097216, MH107182, and MH097216 and R56 AG063433; German Research Foundation (DFG) Postdoctoral Research Fellowship SCHU2710/1-1; and NS064091 and NS039444.