Northwestern Medicine investigators have discovered how a gene mutation results in buildup of a toxic compound known to cause Parkinson’s disease symptoms, defining for the first time the mechanism underlying that aspect of the disease.
The study, published in Neuron, points to a potential novel therapeutic pathway using drugs originally intended to treat another condition, Gaucher’s disease, according to senior author Joseph Mazzulli, PhD, assistant professor in The Ken & Ruth Davee Department of Neurology, Division of Movement Disorders.
In Parkinson’s, a protein called alpha-synuclein is converted to insoluble, toxic clusters in the central nervous system — but the exact mechanism of conversion was unknown until now.
One of the strongest risk factors for developing these clusters is a mutation in the gene GBA1, which normally degrades a lipid called glucosylceramide. Patients with one mutation in the gene suffer an increased level of glucosylceramide and have been shown to have a five-fold increased risk for Parkinson’s; patients with two mutant forms of the gene, one inherited from each parent, can develop Gaucher’s disease, a lysosomal storage disorder.
Mazzulli and his colleagues used this genetic connection to elucidate the mechanism of GBA1-influenced Parkinson’s disease. Using stem cell models of neurons, they used a pharmacological inhibitor to increase levels of glucosylceramide without a mutated GBA1 gene. Even without the mutation, this resulted in a dramatic build-up of toxic alpha-synuclein in neurons.
“This suggested to us that the critical factor in converting alpha-synuclein from its normal form to its pathogenic form was not necessarily the presence of the mutated GBA1 protein, but more importantly the decreased activity and accumulation of glucosylceramide,” Mazzulli said.
With that realization in mind, the investigators took a closer look at the conversion process, finding the healthy form of alpha-synuclein actually existed in two different forms, a simple molecule and a more complex molecule. While it was assumed previously that only the simple molecule was converted into the toxic variety, Mazzulli and his colleagues unexpectedly found the complex molecule was directly converted by glucosylceramide into toxic alpha-synuclein.
“We were surprised to find that toxic aggregation occurred by direct conversion of the large alpha-synuclein complex,” Mazzulli said. “We thought the complex would have to first disassemble before forming toxic aggregates, but that’s not what our data indicated.”
These discoveries suggest that future therapies targeting this pathway might utilize drugs originally intended for treating Gaucher’s disease, Mazzulli said. While the pharmaceutical industry has been interested in the use of those lipid-reducing agents for some time, this study defines the molecular process behind those efforts and demonstrates how it might work.
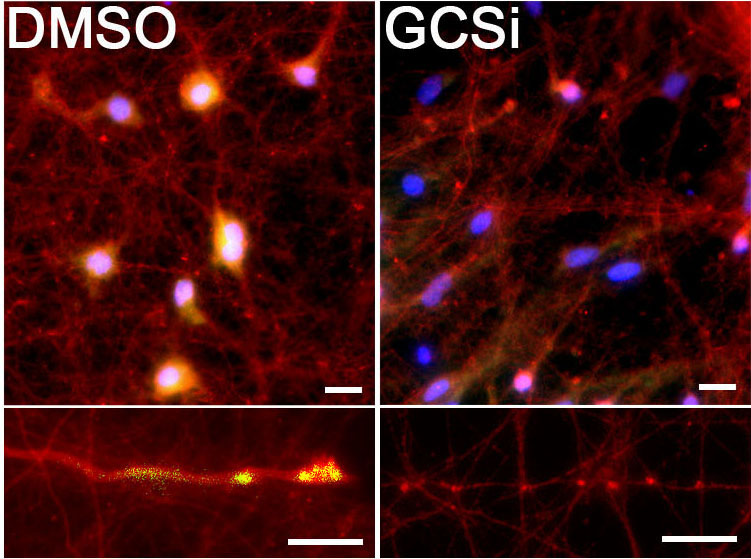
“Some companies have been using synthase inhibitors to reduce the synthesis of the lipid, and we used a similar compound on patient-derived neurons in our study,” Mazzulli said. “We were able to show it reduced toxic alpha-synuclein aggregation directly within neurons derived from Parkinson’s patients.”
In addition, the study gives future drug trials an important metric to measure success, Mazzulli said.
“Our ultimate goal is to reduce alpha-synuclein levels in patients, however measuring the levels of alpha synuclein from the central nervous system of a living patient is complicated,” Mazzulli said. “It’s far easier to measure the effects of therapeutics that alter glucosylceramide in patients, since the lipid can be directly measured from easily accessible fluids, such as blood or cerebral spinal fluid.”
The next step towards a functional treatment will be to graduate from stem cell-derived neurons to animal models, according to Mazzulli.
“We’re trying to determine if this occurs in a living animal and if we can reverse it using similar compounds to what’s being tested in these clinical trials,” Mazzulli said. “If we can intervene before any insoluble amyloid protein develops, maybe we can reverse it to its normal state.”
The first author was Friederike Zunke, PhD, a former postdoctoral fellow in the Mazzulli lab, who has now moved on to lead her own group at the University of Kiel in Germany.
This study was supported by the National Institute of Neurological Disorders and Stroke grant R01NS092823, Northwestern PDMD Advisory Council, the Michael J. Fox Foundation grant ID 12158, and the Human Embryonic and Induced Pluripotent Stem Cell Facility at Northwestern.