Selective Gene Delivery Modifies Diseased Cells
Scientists have demonstrated for the first time that it is possible to specifically modify gene expression in diseased upper motor neurons, brain cells that break down in ALS.
The new Northwestern Medicine study, published in Nature Gene Therapy, provides evidence that lays a foundation for developing future gene replacement therapies to treat patients with the fatal neuromuscular disorder.
Using a nontoxic virus injected directly into the motor cortex of mouse models with ALS, the scientists showed they can deliver new genes to damaged upper motor neurons. This process of transferring DNA from a virus to neurons is called transduction.
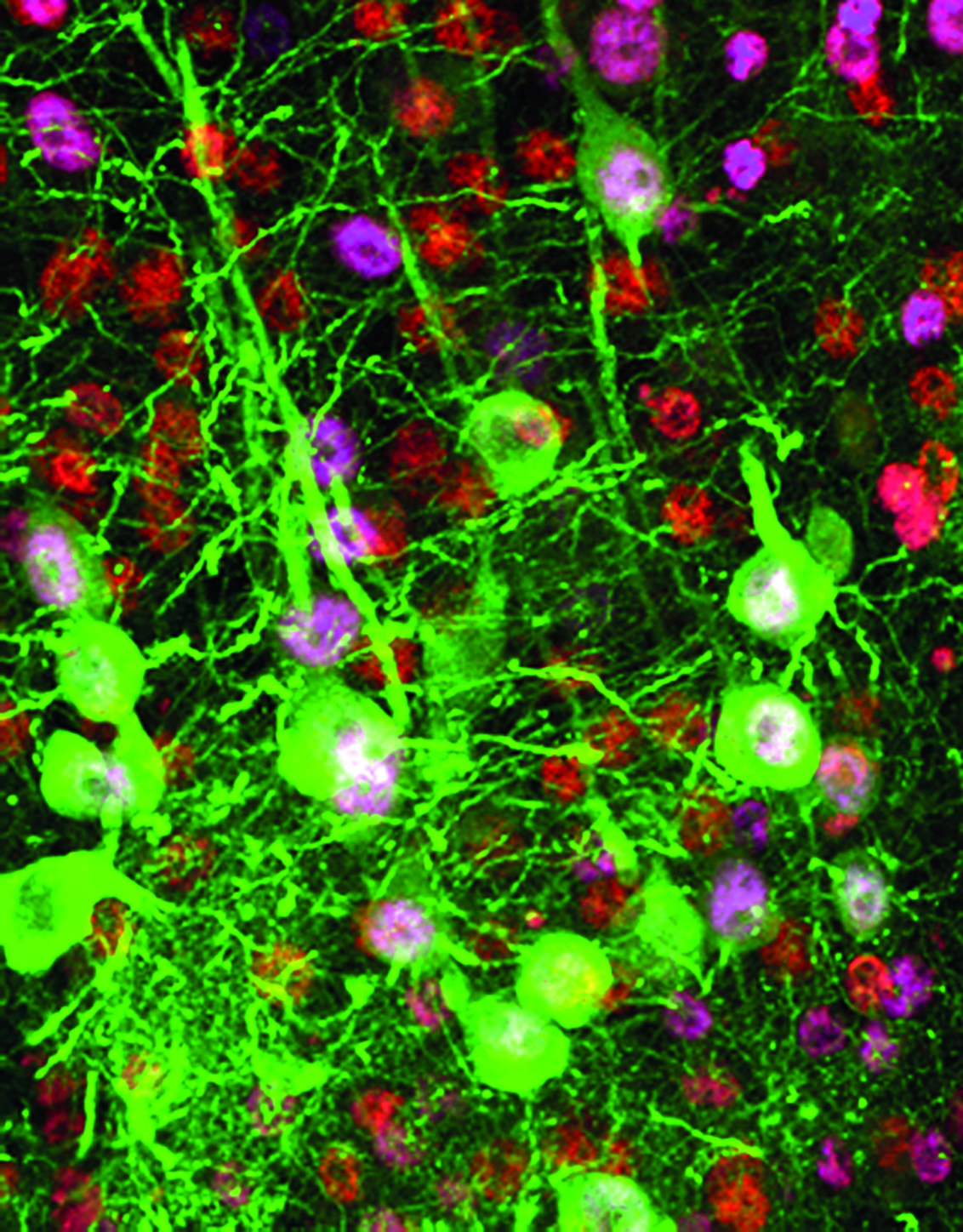
To test the feasibility of transduction, the research team had the virus deliver a gene that expresses a green fluorescent protein – the color helped the scientists visualize how the neurons worked. Now that they know the transduction strategy is effective, they’ll use it in future research to deliver genes that correct mutations in ALS cells.
Importantly, the scientists were able to specifically modify the gene expression in diseased upper motor neurons without disturbing other neurons in the motor cortex. Inadvertently manipulating other cells could set off a cascade of unknown effects.
“The brain is very complex, with many different cells, but in ALS only a distinct neuron population shows initial vulnerability and undergoes progressive degeneration,” said lead study author Hande Ozdinler, PhD, assistant professor of Neurology. “To develop effective treatment strategies, we must deliver genes only to the neurons in need. This is not easy to accomplish – previous studies have managed to induce a broad but non-specific transduction of many different neurons.”
ALS, or amyotrophic lateral sclerosis, is marked by the deterioration of motor neurons, which causes muscle weakness and impaired speaking, swallowing and breathing, eventually leading to paralysis and death. In previous research, Ozdinler showed that defects in upper motor neurons (also known as corticospinal motor neurons), which send messages from the brain to the spinal cord to activate voluntary movement, may be a starting point for the disease.

In the recent study, Ozdinler’s team tested seven different strains of the adeno-associated virus. They found that a nontoxic strain called AAV2-2, already used in clinical studies for diseases such as Parkinson’s, was able to deliver genes to damaged upper motor neurons at rates higher than ever seen before.
“With just a one-time injection into the motor cortex, genes were very specifically delivered to the upper motor neurons,” Ozdinler said. “Among all cells transduced, about 70 percent were upper motor neurons. Without selectivity, this would be about 1 percent.”
The investigators showed that the AAV2-2 virus transduced upper motor neurons in models of both pre-symptomatic and symptomatic stages of ALS, suggesting patients could eventually benefit from therapy based on this work even after they’ve started experiencing symptoms.
“This new study has very important clinical implications, especially for patients with familial ALS who display upper motor neuron defects,” Ozdinler said.
An upper motor neuron projects information to the spinal cord through its axon, a long, branch-like part of the cell that sends impulses to other cells. The fluorescent protein that was transferred to the neurons made this process visible.
“We saw that during ALS, damaged upper motor neurons stop talking to spinal neurons,” Ozdinler said. “In further research, we will examine how we can modulate gene expression to introduce correct versions of mutated genes and improve that connectivity and motor functions.”
Others authors of the study include Javier Jara, PhD, research assistant professor of Neurology, Macdonell Stanford, Yongling Zhu, PhD, assistant professor of Ophthalmology, Michael Tu, William Hauswirth, PhD, Martha Bohn, PhD, professor emeritus of Pediatrics, and Steven DeVries, MD, PhD, professor of Ophthalmology and Physiology.
The research was funded by the Les Turner ALS Foundation, Herbert C Wenske Foundation, Northwestern University Clinical and Translational Sciences (NUCATS) Institute, an ALSA Safenowitz fellowship and the Weinberg College of Arts and Sciences at Northwestern University.
Shedding Light on Cellular Degeneration in Juvenile ALS
In a second recent paper, published in Human Molecular Genetics, Ozdinler revealed novel upper motor neuron defects in a model of the most severe form of juvenile ALS.
Using a fluorescent reporter line developed by Ozdinler’s lab in 2013, the research team genetically labeled the upper motor neurons in mice without the ALS2 gene, which is mutated in cases of early-onset ALS.
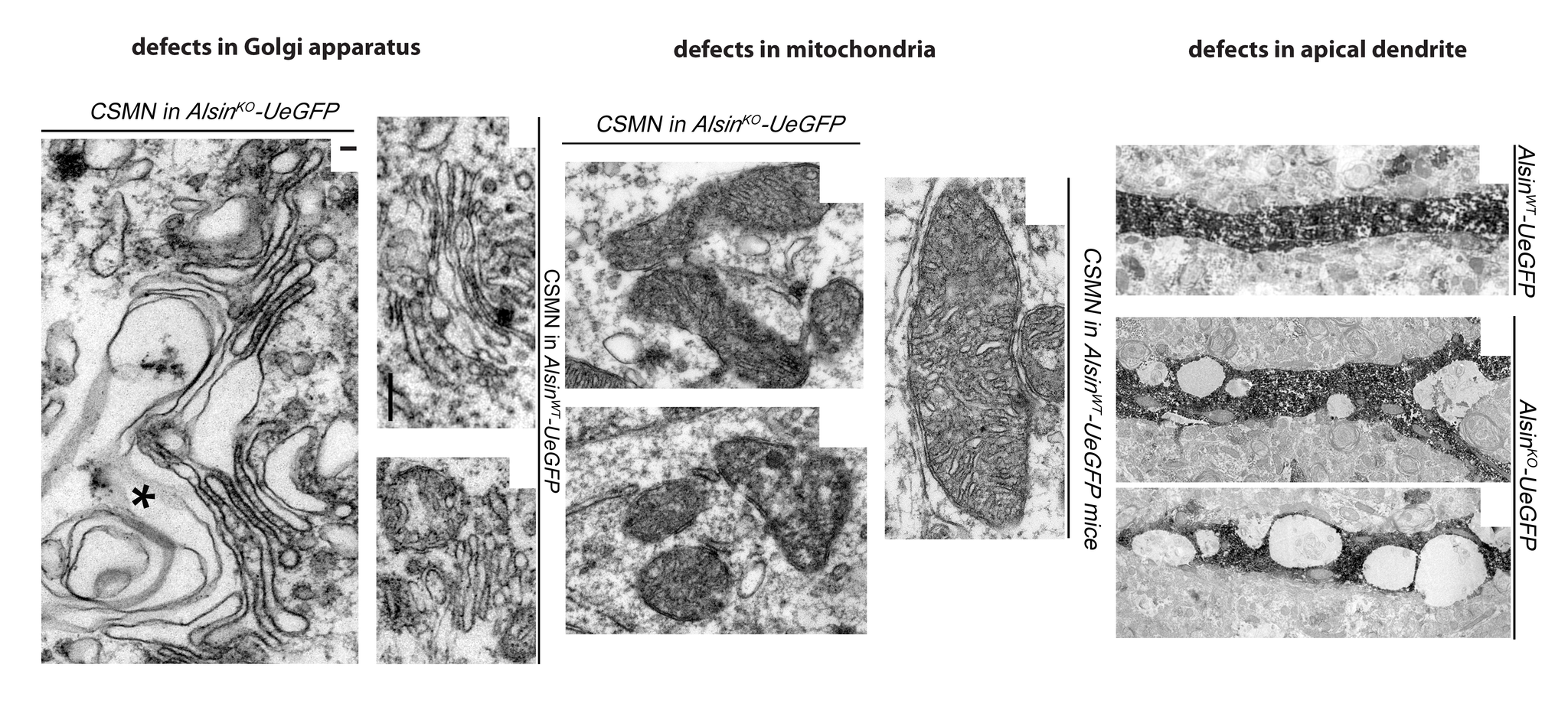
The study showed that vulnerable upper motor neurons display defects in their mitochondria and Golgi apparatus, important functional parts of the cells.
“Revealing novel cellular defects in upper motor neurons is important to develop new drug targets for ALS,” Ozdinler said. “Previously we did not know why ALS patients with the alsin mutation develop the disease so early. Our studies reveal that upper motor neurons displayed defects in their mitochondria, Golgi apparatus and failed to maintain their cellular integrity. This broad spectrum of neuronal dysfunction could indeed cause an early and profound phenotype.”
Mukesh Gautam, PhD, Marco Martina, MD, PhD, associate professor of Physiology, Javier Jara, PhD, and Gabriella Sekerka, MVDr, CSc, ’01 GME, research assistant professor of Physiology, were additional authors of the paper. Han-Xiang Deng, MD, PhD, research professor of Neurology, and Teepu Siddique, MD, professor of Neurology, shared the mouse model that enabled this work. The research was funded by National Institutes of Health grant R21-NS085750-01 and the Les Turner ALS Foundation.